
Abstract
The object of this work was to measure the complete chloroplast genome of Phoebe hunanensis Hand.-Mazz. for the sake of offering valuable genomic information to promote its conservation. The complete chloroplast genome sequence of P. hunanensis was measured as 152,791 bp in size. It contains the typical structure and gene content of angiosperm plastome, which includes a large single-copy (LSC), a small single-copy (SSC), and two inverted repeat (IR) regions of 93,713, 18,928, and 20,075 bp, respectively. The P. hunanensis chloroplast genome has a total of 126 genes including 82 protein-coding genes, 36 tRNA genes, and 8 rRNA genes. Overall GC contents of the genome were 39.15%. Phylogenetic analysis based on the 20 chloroplast genomes measurement demonstrated that P. hunanensis is most closely related to Phoebe bournei.
Phoebe hunanensis Hand.-Mazz. is an outstanding member of Lauraceae family, mainly distributed in subtropical lowland areas with elevations around 500 m. It possesses wide ecological amplitude and strong ability of germination and regeneration. Currently, P. hunanensis possesses high value for timber, oil, medicinal, and ornamental purposes. Its utilization value has been gradually recognized, and it will be widely used in the future. But to date, there is still no complete cp genome was characterized for P. hunanensis. Here, we characterized the complete cp genome sequence of P. hunanensis (GeneBank accession number: MT246867) based on Illumina pair-end sequencing to provide a valuable complete cp genomic resource.
The fresh leaves of P. hunanensis natively habitated on Zijinshan (N 32.053 and E 118.857) in Nanjing, China were collected for whole genome DNA extraction. The voucher specimen was deposited in the herbarium of Nanjing Forestry University (accession number: NF2018797). In this study, we determined the complete chloroplast DNA sequence of P. hunanensis by using next-generation sequencing technology. The total DNA extracting and the whole-genome sequencing were conducted by Nanjing Genepioneer Biotechnologies Inc. (Nanjing, China) with the Illumina Hiseq 2500 Sequencing System. The original reads were filtered by CLC Genomics Workbench v9, and the obtained clean reads were assembled into chloroplast genome using SPAdes assembler version 3.10.1 (Bankevich et al. Citation2012). Finally, CpGAVAS (Liu et al. Citation2012) was used to annotate the gene structure and OGDRAW (Lohse et al. Citation2013) was used to generate the physical map.
The complete cp genome size of P. hunanensis was 152,791 bp, which was composed of a large single-copy region (LSC) of 93,713 bp, a small single-copy region (SSC) of 18,928 bp, and two inverted repeat (IR) regions of 20,075 bp. In total, the genome contained 126 genes, including 82 protein-codon genes, 36 tRNA genes, and 8 rRNA genes. Among them, 15 genes occur in double copies, including 4 protein-codon genes (ndhB, rps12, rps7, and ycf1), 7 tRNA genes (trnA-UGC, trnG-UCC, trnI-GAU, trnL-CAA, trnN-GUU, trnR-ACG, and trnV-GAC), and 4 rRNA genes (16S, 23S, 4.5S, and 5S), and most of the gene occur as a single copy. A total of eight protein-coding genes (ndhA, ndhB, petB, petD, rpl16, rpl2, rpoC1, and rps16) contained one intron while the other three genes (clpP, rps12, and ycf3) had two introns each. The whole chloroplast genome GC content is 39.15%, while the value of the LSC, SSC, and IR regions are 37.95, 33.84, and 44.43%, respectively.
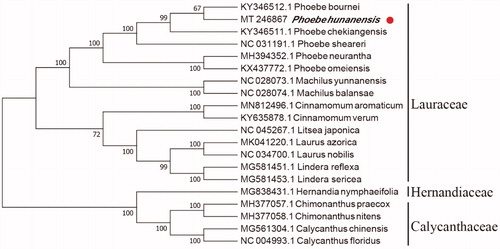
In order to reveal the phylogenetic position of P. hunanensis with other members of Lauraceae, a phylogenetic analysis was performed based on 15 complete chloroplast genomes of Lauraceae, and two taxa (Calycanthaceae and Hernandiaceae) as outgroups. They were all downloaded from NCBI GenBank. Based on the neighbor-joining (NJ), the phylogenetic tree was deduced by MAFFT version 7.307 (Katoh and Standley Citation2013) and MEGA version 7 (Kumar et al. Citation2016). As shown in , P. hunanensis is most related to Phoebe bournei, with bootstrap support values of 67%.
Figure 1. Phylogenetic tree inferred by maximum-likelihood (ML) method based on the complete chloroplast genome of 20 representative species. The bootstrap support values are shown at the branches.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability
The data is accessible from:
https://pan.baidu.com/s/1fFE37q1pC8-GJuLSqI7YMw (password: akwm) https://pan.baidu.com/s/1W5tlSTupHAYPl-QXASG9AA (password: un07)
Additional information
Funding
References
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, Pyshkin AV, et al. 2012. SPAdes: new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 19(5):455–477.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability (Article). Mol Biol Evol. 30(4):772–780.
- Kumar S, Stecher G, Tamura K. 2016. Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 33(7):1870–1874.
- Liu C, Shi LC, Zhu YJ, Chen HM, Zhang JH, Lin XH, Guan XJ. 2012. CpGAVAS, an integrated web server for the annotation, visualization, analysis, and GenBank submission of completely sequenced chloroplast genome sequences. BMC Genomics. 13(1):715.
- Lohse M, Drechsel O, Kahlau S, Bock R. 2013. Organellar Genome DRAW—a suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 41(W1):W575–W581.