
Abstract
Camellia amplexicaulis (Pit.) Cohen-Stuart is a shrub or small tree that extensively cultivated throughout Vietnam and beyond, which is now considered to extinct in the wild. Here, the whole plastid genome of C. amplexicaulis was assembled using Illumina paired-end sequencing reads. The plastid genome is 157,203 bp in length with a typical quadripartite structure, containing two copies of inverted repeat (IR) regions (26,133 bp), a large-single copy (LSC) region (86,644 bp), and a small-single copy (SSC) region (18,293 bp). A total of 114 unique genes were encoded, of which 80 are protein-coding genes, 30 are tRNA genes, and four are rRNA genes. The phylogenetic result indicates C. amplexicaulis is closely related to Camellia szechuanensis C. W. Chi.
Camellia L., comprising ca. 120 species, is the type and the economically and phylogenetically important genus in the family Theaceae (Vijayan et al. Citation2009; Yu et al. Citation2017). Species of the genus is mostly distributed in southeastern and eastern Asia, 82% of which inhabit the subtropical (the subtropical evergreen broadleaved forests) EBLFs of East Asia (Yu et al. Citation2017). Camellia amplexicaulis (Pit.) Cohen-Stuart is a shrub or small tree that extensively cultivated throughout Vietnam and beyond (Rivers 2018). The species was originally found in the Tam Dao National Park in North Vietnam and is now considered to extinct in the wild (Rivers Citation2018). In this study, we characterized the whole chloroplast genome of C. amplexicaulis. Total genome DNA was extracted from fresh leaves of an individual of C. amplexicaulis using a modified CTAB protocol (Doyle and Doyle Citation1987), which collected from South China Botanical Garden and deposited at South China Botanical Garden Herbarium (voucher number: Wu19016).
The Illumina paired-end (2 × 150 bp) library was prepared and sequenced at Beijing Genomics Institute (Wuhan, China) based on Illumina Hiseq X Ten platform. Clean data were obtained after removing low quality reads and adaptor sequences. The complete plastid genome of C. amplexicaulis was assembled using NOVOPlasty (Dierckxsens et al. Citation2017) with a reference plastid genome C. granthamiana (NC_038181). We then rechecked the draft plastid genome by remapping reads in Geneious Prime 2019 (Biomatters, Ltd, Auckland, New Zealand). The coding and tRNA genes in the plastid genome were annotated using Geneious and ARAGORN (Laslett and Canback Citation2004), respectively.
The plastome of C. amplexicaulis (GenBank accession number MT317095) is 157,203 bp in length with a typical quadripartite structure, containing two copies of inverted repeat (IR) regions (26,133 bp), a large-single copy (LSC) region (86,644 bp), and a small-single copy (SSC) region (18,293 bp). The overall GC content of the genome is 37.3%. Specifically, the GC values of the LSC, SSC, and IR regions are 35.3, 30.5, and 42.9%, respectively. A total of 114 unique genes were encoded, of which 80 are protein-coding genes, 30 are tRNA genes, and four are rRNA genes.
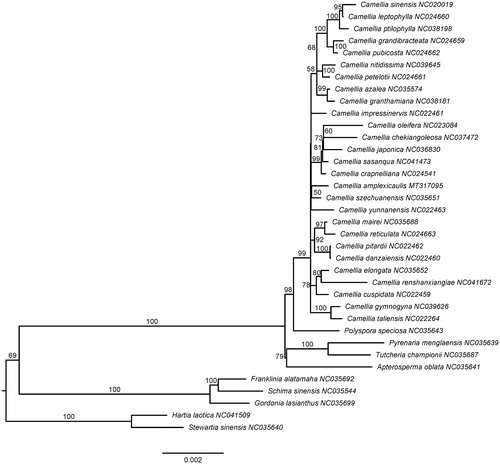
Plastid genomes of 26 species in Camellia and 9 outgroups from different close-relative genera in Theaceae were downloaded from GenBank. The aligned matrix comprised of 78 protein-coding genes extracted from 36 whole plastid genomes was implemented in MAFFT (Katoh and Standley Citation2013). The maximum-likelihood tree was constructed using RAxML (Stamatakis Citation2014) with 1000 rapid bootstrap replicates. The ML tree showed the species of Camellia formed a monophyletic clade with 99% bootstrap support value (). After the lineage deriving from the recent common ancestor of Camellia, a polytomy topology was found at the intrageneric level. C. amplexicaulis and Camellia szechuanensis C. W. Chi were recovered as sister relationship, however, without solid support by bootstrap (50%).
Figure 1. Phylogenetic relationships of Camellia inferred from maximum likelihood method based on 78 common protein-coding genes. The node labels indicate the ML bootstrap (1000 replicates) support values.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability
The data that support the findings of this study are openly available in GenBank at https://www.ncbi.nlm.nih.gov/, reference number MT317095.
Additional information
Funding
References
- Dierckxsens N, Mardulyn P, Smits G. 2017. NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 45(4):e18.
- Doyle JJ, Doyle JL. 1987. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull. 19:11–15.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30(4):772–780.
- Laslett D, Canback B. 2004. ARAGORN, a program for the detection of transfer RNA and transfer-messenger RNA genes in nucleotide sequences. Nucleic Acids Res. 32(1):11–16.
- Rivers MC. 2018. Camellia amplexicaulis. The IUCN Red List of Threatened Species 2018. e.T191323A1975870. http://dx.doi.org/10.2305/IUCN.UK.2018-1.RLTS.T191323A1975870.en.
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30(9):1312–1313.
- Vijayan K, Zhang WJ, Tsou CH. 2009. Molecular taxonomy of Camellia (Theaceae) inferred from nrITS sequences. Am J Bot. 96(7):1348–1360.
- Yu XQ, Gao LM, Soltis DE, Soltis PS, Yang JB, Fang L, Yang SX, Li DZ. 2017. Insights into the historical assembly of East Asian subtropical evergreen broadleaved forests revealed by the temporal history of the tea family. New Phytol. 215(3):1235–1248.