
Abstract
Begonia versicolor (Code: MK434325) is a perennial plant with unique and precious ornamental value, exhibiting gorgeous leaf morphology and graceful flowers, therefore, it is popular with many horticulturalists and plant hobbyist. The complete chloroplast genome of B. versicolor were reported in this article. It had a typical quarter structure as a circular of 169,506 bp, composed by a large single-copy region (LSC, 75,868 bp), a small single-copy region (SSC, 18,290 bp), and two inverted repeats (IRs, 75,258 bp each). Genome annotations showed that it contained total 185 genes, including 133 protein coding genes (PCGs), 44 transfer RNA (tRNAs), and 8 ribosome RNA (rRNAs). The percentage of total GC content was 35.57%. Phylogenetic analysis suggested that Begoniaceae was closely related to Cucurbitaceae family.
Begonia L., the sixth largest genus in the world, belongs to Begonaceae family, which consists of about 1500 accepted species. They are distributed in tropical and subtropical regions of the America, Asia, and Africa between 30°N and 30°S. It has been worldwidely applied for greening and decoration as acclaimed landscape plants for a long times. China is one of the most suitable habitat for Begonia species with over 200 species, what is more, its extraordinarily abundant wild plant resources are mainly contained by Yunnan province (southwest China) (Doorenbos et al. Citation1998; DeWitt et al. Citation2011). Begonia versicolor is an endemic species in China, with gorgeous leaf ornamental and graceful flowers, and it is favored by breeders and horticulturalists.
Currently, although there are many different methods to identify Begonia species through morphological traits, such as leaf, flower, fruit, etc., with its wide morphology variations, there is still a lot of controversy among different taxonomists. In addition, the genetic information is supplied limitedly in most begonias, which makes it difficult on phylogenetic research. Chinese begonias often grow in shady and moist environment, however, due to the impact of human activities and global climate change, many Begonia species have faced serious problems of habitat destruction in recent years (Guan et al. Citation2008). Therefore, we hope to further resolve infrageneric relationships among Begonia by sequencing the chloroplast genome, and provide a foundation for in-depth exploration of the phylogenetic history of Begonia and its biodiversity protection.
The plant samples of B. versicolor were collected from Yunnan Province, China (104°37′51′′ E; 23°6′10′′ N). The specimen (ID: EM0517) was deposited in the herbarium of Sun Yat-sen University (SYS). The extracted genomic DNA was sequenced base on Illumian platform of whole genome pair-end sequencing. The complete chloroplast genome was assembled and annotated by GetOrganelle (Jin et al. Citation2018) and Plann (Huang and Cronk Citation2015). The genome size of B. versicolor was 169,506 bp (GenBank accession ID: MK434325), which contained a large single-copy region (LSC, 65,868 bp), a short single-copy region (SSC, 18,290 bp) and two inverted repeat sequences (IRs, 75,258 bp, each). The chloroplast circle was totally comprised of 185 genes (141 unique genes), including 133 protein-coding genes (PCGs, 108 unique genes), 8 ribosomal RNA (rRNAs, 4 unique genes), and 44 transfer RNA (tRNAs, 29 unique genes). The total GC content of the whole chloroplast genome was 35.57%.
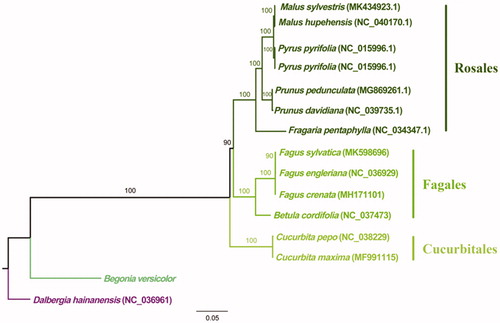
In order to confirm the phylogenetic position of B. versicolor. All the 15 sequences (14 downloaded from NCBI) were aligned via MAFFT v.7.271 (Katoh and Standley Citation2013) and jmodeltest v.2.1.10 (David Citation2008) was applied for model test. Eventually the phylogenetic maximum likelihood (ML) tree was structured by means of RAxML v.8.2.12 (Stamatakis Citation2014). As a result, the phylogenetic tree () strongly supported that the B. versicolor formed a separate branch and has a closer relationship with Cucurbita (Cucurbitaceae). This result is consistent with phylogenetic relationship between Cucurbitales order in the latest classification of flowering plants (APG IV).
Figure 1. Maximum-likelihood tree based on complete chloroplast genomes. Bootstrap support values are shown next to the nodes.
Acknowledgment
We thank Yanzhao Chen and Yubing Zhou for assistance in data processing.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The data that support the findings of this study are available in NCBI at https://www.ncbi.nlm.nih.gov/, reference number [MK434325].
Additional information
Funding
References
- David P. 2008. jModelTest: phylogenetic model averaging. Mol Biol Evol. 25(7):1253–1256.
- DeWitt A, Twyford A, Thomas D, Kidner C, Van Huylenbroeck J. 2011. The origin of diversity in Begonia: genome dynamism, population processes and phylogenetic patterns. In: The dynamical processes of biodiversity-case studies of evolution and spatial distribution. The dynamical processes of biodiversity: evolution and spatial distribution. Few study cases; p. 27–52.
- Doorenbos J, Sosef MSM, de Wilde J. 1998. The sections of Begonia including descriptions, keys and species lists (Studies in Begoniaceae VI). Wageningen (The Netherlands): Wageningen Agricultural University. Wageningen Agricultural University papers; No. 98–2.
- Guan KY, Ma H, Li JX, Li HZ, Yamaguchi H. 2008. Begonia germplasm resources of China. Acta Hortic. 766(766):337–348.
- Huang DI, Cronk Q. 2015. Plann: a command-line application for annotating plastome sequences. Appl Plant Sci. 3(8):1500026.
- Jin JJ, Yu WB, Yang JB, Song Y, Yi TS, Li DZ. 2018. GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. BioRxiv. 256479. DOI: 10.1101/256479.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30(4):772–780.
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30(9):1312–1313.