
Abstract
The complete mitogenome of Saccharum spp. hybrid FN15 was successfully sequenced. It contains two distinct circular chromosomes, Chromosome 1 and Chromosome 2. The former is 301,533 bp in length with the GC content of 43.90%, and 7.12% of genome (21,468 nucleotides) are coding DNA while 92.88% of genome (280,065 nucleotides) are intergenic region. The latter is 144,744 bp in length with the GC content of 43.57%, and 8.20% of genome (11,865 nucleotides) are coding DNA and 91.80% of genome (132,879 nucleotides) are intergenic region. Besides, Chromosome 1 contains 22 protein-coding genes (four atp genes, three ccm genes, three cox genes, one mat gene, one mtt gene, six nad genes and four rps genes), and 21 non-coding genes (15 tRNA and six rRNAs), whereas in Chromosome 2, there are 13 protein-coding genes (two atp genes, one ccm gene, one cob gene, one cox gene, one rpl gene, four nad genes and three rps genes) and five tRNA genes. Maximum Likelihood phylogenetic analysis demonstrated that FN15 is close with S. spp. hybrid ROC22, S. officinarum Khon Kaen 3 and S. bicolor species. This complete mitochondrial genome will provide essential DNA molecular data for further phylogenetic and evolutionary analysis for Saccharum.
Sugarcane (Saccharum spp.), which belongs to the family of Gramineae, accounts for ∼80% of sugar production and ∼60% of bioethanol production worldwide and therefore is the most important sugar and bioenergy crop (Garsmeur et al. Citation2018; Zhou et al., Citation2020). Modern commercial sugarcane varieties are all complex interspecies hybrids (Garsmeur et al. Citation2018; Zhou et al. Citation2018). FN15, a Saccharum spp. hybrid from CP72-1210 × YN73-204, is a national identified commercial variety and is approved to be planted in main sugarcane production provinces since 2005 in China (Zhang and Govindaraju Citation2018) and serves as one of the important crossing parents. The characterization of the complete mitogenome of sugarcane variety FN15 and its phylogenetic relationship within Gramineae were described in the present study.
The sugarcane variety FN15 was sampled from Fujian Agriculture and Forestry University, Fuzhou, Fujian Province (geographic coordinates: 26°9′8″N, 119°24′24″E), China. The specimen of sugarcane FN15 was stored in the Key Laboratory of Sugarcane Biology and Genetic Breeding, Fujian Agriculture and Forestry University with store number FN15- FJ2016001. An improved protocol, which reported by Chen et al. (Citation2011), was firstly adopted to extract and purify the mitochondrial DNA (mtDNA) from fresh yellowing seedlings. Then, the complete mitochondrial genome was sequenced using Illumina Hiseq XTen and PacBio Sequel platform, and assembled by SPAdes v3.10.1 (Antipov et al. Citation2016). Thereafter, the genome sequences were annotated using GeSeq (Tillich et al. Citation2017). The complete mitochondrial genome sequence has been submitted to GenBank with the accession numbers: MT411890 (Chromosome 1) and MT411891 (Chromosome 2).
The complete mitogenome of sugarcane variety FN15 includes two distinct annular chromosomes, termed as Chromosome 1 and Chromosome 2. The Chromosome 1 is 301,533 bp in length with the GC content of 43.90%, and 7.12% of genome (21,468 nucleotides) are coding DNA while 92.88% of genome (280,065 nucleotides) are intergenic region. The latter is 144,744 bp in length with the GC content of 43.57%, and 8.20% of genome (11,865 nucleotides) are coding DNA and 91.80% of genome (132,879 nucleotides) are intergenic region. Interestingly, Chromosome 1 contains 22 PCGs (protein-coding genes, four atp genes, three ccm genes, three cox genes, one mat gene, one mtt gene, six nad genes and four rps genes), and 15 tRNAs and six rRNAs non-coding genes, whereas Chromosome 2 includes five tRNA genes and 13 PCGs (four nad genes, three rps genes, two atp genes, one ccm gene, one cob gene, one cox gene, and one rpl gene). Regarding the initiation and stop codon, all these PCGs in Chromosome 1 use the initiation codon ATG except for nad1 and matR, which start with ACG and ATA, respectively, and most of them terminate with TGA, TAG, or TAA, whereas nad2 and nad5 stop with CGG and GTA, respectively. However, in Chromosome 2, all these PCGs begin with ATG except for nad2 and nad5, which start with TTG and CCA, respectively, and ccmFc, rps12, cox3, and nad4 terminate with TGA, whereas atp9, rps3, and cob terminate with TAG, and the other genes (atp6, nad3, rps4, rpl16, nad2, and nad5) terminate with TAA.
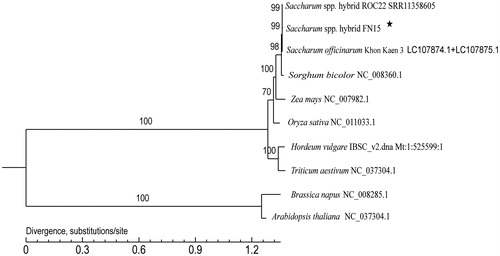
To explore the phylogenetic position of Saccharum spp. hybrid FN15, a phylogenetic tree by Maximum-Likelihood with 1000 bootstrap replications was constructed based on the whole mitochondrial genomes of 10 species using PhyML v3.0 (http://www.atgc-montpellier.fr/phyml/). GenBank accession numbers for each plant species are as follows: Saccharum spp. hybrid ROC22 (SRR11358604), Oryza sativa (NC_011033.1), Zea mays (NC_007982.1), Sorghum bicolor (NC_008360.1), Triticum aestivum (NC_037304.1), Hordeum vulgare (IBSC_v2.dna. Mt: 1:525599:1 REF), Saccharum officinarum Khon Kaen 3 (LC107874.1 and LC107875.1), Arabidopisis thaliana (NC_037304.1), and Brassica napus (NC_008285.1), with the last two species used as outgroups. The result indicated that the phylogenetic relationship of Saccharum spp. hybrid FN15 is very close to S. spp. hybrid ROC22 (99%), S. officinarum Khon Kaen 3 (99%), and S. bicolor (98%), which all belong to the species of family Gramineae (). The complete mitochondrial genome sequence will provide essential and important DNA molecular data for phylogenetic and evolutionary analysis for Saccharum. Significant DNA molecular data offered by the complete mitochondrial genome sequence herein will contribute to further phylogenetic and evolutionary analysis of Saccharum.
Figure 1. A maximum-likelihood phylogenetic tree constructed based on the comparison of mitochondrial genome sequences of ten species. GenBank accession numbers are listed after the species name. The numbers at the nodes are bootstrap percent probability values based on 1000 replications. The genome sequence in this study is labeled with an asterisk.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The data that support the findings of this study are available. The mitochondrial genome sequences of analyzed species (Saccharum spp. hybrid FN15, O. sativa, Z. mays, S. bicolor, T. aestivum, S. officinarum Khon Kaen 3, A. thaliana, and B. napus) were from the GenBank databases (https://www.ncbi.nlm.nih.gov/), Saccharum spp. hybrid ROC22 mitchondrial genome is available in NCBI SRA with the accession number of SRR11358605 (https://dataview.ncbi.nlm.nih.gov/objects?linked_to_id=SRR11358605&archive=biosample), and the mitogenome of H. vulgare is available in ensembl (http://asia.ensembl.org/index.html).
Additional information
Funding
References
- Antipov D, Korobeynikov A, McLean JS, Pevzner PA. 2016. hybridSPAdes: an algorithm for hybrid assembly of short and long reads. Bioinformatics. 32(7):1009–1015.
- Chen J, Guan R, Chang S, Du T, Zhang H, Xing H. 2011. Substoichiometrically different mitotypes coexist in mitochondrial genomes of Brassica napus L. PLoS One. 6(3):e17662
- Garsmeur O, Droc G, Antonise R, Grimwood J, Potier B, Aitken K, Jenkins J, Martin G, Charron C, Hervouet C, et al. 2018. A mosaic monoploid reference sequence for the highly complex genome of sugarcane. Nat Commun. 9(1):2638
- Tillich M, Lehwark P, Pellizzer T, Ulbricht-Jones ES, Fischer A, Bock R, Greiner S. 2017. GeSeq - versatile and accurate annotation of organelle genomes . Nucleic Acids Res. 45(W1):W6–W11.
- Zhang M, Govindaraju M. 2018. Sugarcane production in China. In de Oliveira AB, editor. Sugarcane-technology and research. London: IntechOpen.
- Zhou D, Yin Z, Liu X, Li Z, Yan M, Que Y, Xu L. 2020. The complete mitochondrial genome sequence and phylogenetic analysis of sugarcane (Saccharum spp.) cultivar ROC22. Mitochondrial DNA Part B. 5(2):1915–1916. doi:10.1080/23802359.2020.1756492.
- Zhou D, Liu X, Gao S, Guo J, Su Y, Ling H, Wang C, Li Z, Xu L, Que Y. 2018. Foreign cry1Ac gene integration and endogenous borer stress-related genes synergistically improve insect resistance in sugarcane. BMC Plant Biol. 18(1):342