
Abstract
Prunus conradinae is a flowering cherry species with high ornamental value. In this study, the complete chloroplast (cp) genome of P. conradinae was obtained using a genome skimming approach. The cp genome was 158,019 bp long, with a large single-copy region of 85,910 bp and a small single-copy region of 19,247 bp separated by two inverted repeats of 26,431 bp. It encodes 130 genes, including 85 protein-coding genes, 37 tRNA genes, and eight ribosomal RNA genes. The phylogenetic analysis indicated that P. conradinae is closely related to the congeners P. maximowiczii, P. takesimensis, P. speciosa, P. serrulata var. spontanea, P. discoidea, and P. matuurai.
Prunus conradinae Koehne, also known as Cerasus conradinae (Koehne) Yü et Li, is a flowering cherry species with high ornamental value. It is mainly distributed in the Shanxi, Henan, Hubei, Hunan, Sichuan, Fujian, and Zhejiang province of China and usually grows in or on the edge of forests and valleys at 500–2100 m altitude, flowers in March (Wu and Raven Citation2003). A good knowledge of genome information and the phylogenetic position of P. conradinae would be essential to the formulation of efficient strategies for its conservation, exploitation, and utilization. To facilitate such purposes, the complete chloroplast (cp) genome of P. conradinae was assembled, and its phylogenetic status in Prunus sensu lato was analyzed.
The fresh leaves were collected from Yongshun county, Hunan province, China (28°47′28′′N, 110°16′36′′E). The voucher specimen (JIU04068) was deposited in the herbarium of Jishou University. Total genomic DNA was extracted by modified CTAB protocol (Doyle and Doyle Citation1987). The whole genome sequencing was performed on the Illumina Hiseq 2500 platform (Illumina, San Diego, CA). A total of 8.2 Gb clean reads were obtained, after removing low-quality reads by fastp software (Chen et al. Citation2018). Then, the clean reads were corrected by BFC tool (Li Citation2015). The processed data was assembled by GetOrganelle pipeline (https://github.com/Kinggerm/GetOrganelle), which exploits Bowtie (Langmead et al. Citation2009), BLAST (Camacho et al. Citation2009), and SPAdes (Bankevich et al. Citation2012) as dependencies. The cp genome was annotated by Dual Organellar GenoMe Annotator (DOGMA) (Wyman et al. Citation2004).
The cp genome of P. conradinae (GenBank accession number: MT374065) was 158,019 bp in length, consisting two inverted repeats (IR), a large single copy (LSC), and a small single copy (SSC), and the sequence lengths are 26,431 bp, 85,910 bp, and 19,247bp, respectively. The overall GC content of the cp genome is 36.7%, while the GC percentages in IR, LSC, and SSC are 42.5%, 34.6%, and 30.1%, respectively. The genome possesses 130 genes, including 85 protein-coding genes, 37 tRNA genes, and eight rRNA genes.
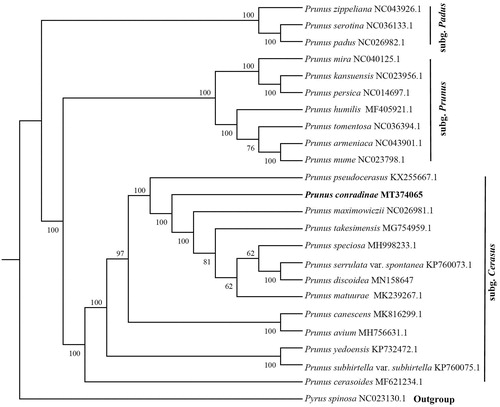
Phylogenetic analysis was conducted to confirm the phylogenetic position of newly sequenced P. conradinae amongst 22 representative P. sensu lato species and an outgroup taxa. We reconstructed a phylogeny employing the GTRGAMMA model and 1000 bootstrap (BS) replicates under the maximum-likelihood (ML) inference in RAxML (Stamatakis Citation2014). The ML tree () showed that all the species were divided into three clades (BS 100%), subg. Cerasus, subg. Prunus, and subg. Padus, which is consistent with previous results (Shi et al. Citation2013), and P. conradinae is closely related to the congeners P. maximowiczii, P. takesimensis, P. speciosa, P. serrulata var. spontanea, P. discoidea, and P. matuurai.
Figure 1. Maximum-likelihood phylogenetic tree for Prunus conradinae based on complete chloroplast genome of 22 taxa of Prunus sensu lato. Pyrus spinosa (Rosaceae) was used as outgroup. The support values are shown at the branches.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The data that support the findings of this study are openly available in GenBank, National Center for Biotechnology Information (NCBI) at https://www.ncbi.nlm.nih.gov/genbank/, with accession number of MT374065.
Additional information
Funding
References
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, et al. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 19(5):455–477.
- Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL. 2009. BLAST+: architecture and applications. BMC Bioinformatics. 10:421.
- Chen S, Zhou Y, Chen Y, Gu J. 2018. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 34(17):i884–i890.
- Doyle JJ, Doyle JL. 1987. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull. 19:11–15.
- Langmead B, Trapnell C, Pop M, Salzberg SL. 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10(3):R25.
- Li H. 2015. BFC: correcting Illumina sequencing errors. Bioinformatics. 31(17):2885–2887.
- Shi S, Li J, Sun J, Yu J, Zhou S. 2013. Phylogeny and classification of Prunus sensu lato (Rosaceae). J Integr Plant Biol. 55(11):1069–1079.
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30(9):1312–1313.
- Wu ZY, Raven PH. 2003. Flora of China: Pittosporaceae through Connaraceae. Beijing: Science Press/St. Louis: Missouri Botanical Garden Press.
- Wyman SK, Jansen RK, Boore JL. 2004. Automatic annotation of organellar genomes with DOGMA. Bioinformatics. 20(17):3252–3255.