
Abstract
In this study, the mitochondrial genome (mitogenome) sequence of Papilio memno is determined using next-generation sequencing. The entire mitogenome genome is determined to be 15,262 bp in length. It contains 37 genes, including 13 protein-coding genes (PCGs), 22 transfer RNA genes (tRNAs), two ribosomal RNA genes (rRNAs), and an adenine (A) + thymine (T)-rich region. The overall GC content of the genome is 20.3%. A phylogenetic tree reconstructed by 39 mitochondrial genomes reveals that P. domestica is most closely related to Prunus salicina.
Papilionidae including about 600 species, are distributed worldwide and many of them have large beautiful wings, have greatly contributed to studies of ecology, behavior and evolution in insects (Häuser et al. Citation2005). The butterfly Papilio memno is one of the most ornamental butterflies and mainly distributed in China, Japan, and South Asian Countries. P. memnon is famous for female-limited Batesian mimicry polymorphism, which is an intriguing system for investigating the mechanism of maintenance of genetic polymorphisms (Komata et al. Citation2016). To date, there is less information about the complete mitochondrial genomes of P. memnon. In our study, the complete mitogenome of P. memnon is determined, which would provide basic genome information for further genetic studies, phylogenetic analysis, and conservation of this species.
Total genomic DNA was extracted from the thorax muscle of a single individual butterfly (N24°30′12.78″, E118°03′14.16″) using the Sangon Animal genome DNA Extraction Kit (Shanghai, China). The voucher specimen was deposited at Southwest Forestry University (MFD: 180705). The whole genome sequencing was conducted by Hefei Biodata Biotechnologies Inc. (Hefei, China) on the Illumina Hiseq 4000 Sequencing System (Illumina, Hayward, CA). The filtered sequences were assembled using the SPAdes assembler 3.10.0 (Bankevich et al.Citation2012). Annotation was performed using the BLAST+ (Camacho et al. Citation2009). and tRNAscan (Schattner et al. Citation2005).
The mitogenome (NCBI acc. no. MH981597) of P. memnon was determined to comprise double stranded, circular DNA of 15,262 bp containing 13 protein-coding genes (PCGs), 22 transfer RNA genes (tRNAs), two ribosomal RNA genes (rRNAs), and an adenine (A) + thymine (T)-rich region with 92.6% AT content. Among these, 14 genes were encoded on the L-strand, including four PCGs (ND1, ND4, ND4Land ND5), two rRNA genes, eight tRNA genes (tRNAGlu, tRNACys, tRNATyr, tRNAPhe, tRNAHis, tRNAPro, tRNAleu and tRNAVal) and A + T-rich region. The remaining 23 genes are encoded on the H strand. The arrangement of genes is similar to all know Papilionidae species. The overall GC content of the genome is 20.3%.
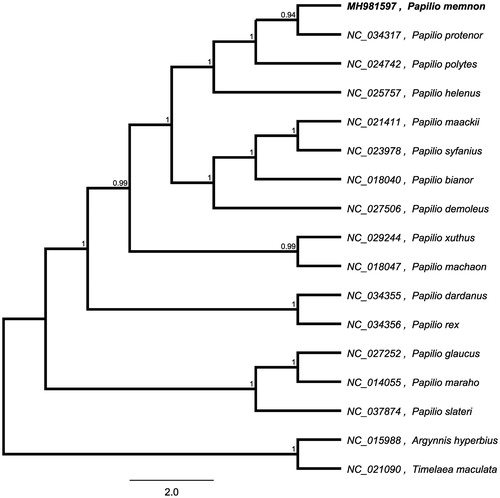
To investigate its taxonomic status, a maximum likelihood (ML) was reconstructed based on whole mitogenome from 15 Papilio butterfly and two outgroup butterfly (Argynnis hyperbius and Timelaea maculata) by Mafft Version 7.0 and FastTree version 2.1.10 (Price Citation2010; Katoh et al. Citation2013). The ML phylogenetic tree shows that P. memnon is most closely related to P. protenor, with bootstrap support values of 100% ().
Figure 1. Maximum-likelihood phylogenetic tree based on whole mitogenome from 15 Papilio buttfly and two outgroup buttfly (Argynnis hyperbius and Timelaea maculata) and the support values are shown at the branches.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability
The complete mitogenome sequence of Papilio memno is deposited in the GenBank database under the accession number MH981597 (https://www.ncbi.nlm.nih.gov/nuccore/MH981597.1/).
Additional information
Funding
References
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, et al. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19(5):455–477.
- Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL. 2009. BLAST+: architecture and applications. BMC bioinformatics. 10(1):421.
- Häuser CL, de Jong R, Lamas G, Robbins RK, Smith C, Vane-Wright RI. 2005. Papilionidae–revised GloBIS/GART species checklist (2nd draft). URL: http://www.insects-online.de/frames/papilio.htm. Accessed December, 2019.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability[J]. Mol. Biol. Evol. 30(4):772–780.
- Komata S, Lin C-P, Iijima T, Fujiwara H, Sota T. 2016. Identification of doublesex alleles associated with the female-limited Batesian mimicry polymorphism in Papilio memnon[J]. Sci Rep. 6:34782.
- Price MN, Dehal PS, Arkin AP. 2010. FastTree 2-approximately maximum-likelihood trees for large alignments . PloS one. 5(3):e9490.
- Schattner P, Brooks AN, Lowe TM. 2005. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs[J]. Nucleic Acids Res. 33(Web Server issue):W686–W689.