
Abstract
The mitochondrial genome (mitogenome) of the Ergatettix serrifemora (Orthoptera: Tetrigidae: Tetriginae) was sequenced and annotated. The assembled mitochondrial genome was 14,947 bp, containing 45.8% of A, 15.7% of C, 9.6% of G and 28.9% of T, respectively, which is the classical structure for insect mitogenome. The region that we failed to sequence was between rrnS and trnI, and generally contained a putative AT-rich region. Twelve PCGs started with typical ATN codon and eleven ended with complete stop codons (three with TAG, eight with TAA). The phylogenetic trees in the current study confirmed that E. serrifemora was clustered with other Tetriginae species, and this study would improve our understanding for the mitogenomes of Tetrigoidea.
The genus Ergatettix belongs to the subfamily Tetriginae, within the family Tetrigidae of the order Orthoptera. This genus currently includes 19 known species, which are mainly distributed in India, Nepal, Myanmar, Philippines, Malaysia, New Guinea, Iran and China (Deng Citation2016). Up to now, no mitochondrial sequence has been reported of the genus. In this study, the partial mitochondrial genome (mitogenome) of E. serrifemora (GenBank accession No. MN938923) was sequenced and determined using next-generation sequencing method for the first time, which will facilitate future studies on species identification, population genetics, and phylogenetic analysis of the subfamily Tetriginae.
Total genomic DNA was extracted from legs of adult specimen of E. serrifemora which was collected from Shanghang county in Fujian province, China, in August 2019 and voucher specimen are deposited in the Museum of Insects of Hechi University, label number is No. O203. The genomic DNA was sequenced using the Hiseq2500 platform (Illumina Inc., San Diego, CA). The mitogenome was assembled using Geneious (Kearse et al. Citation2012), version 10.2.3 (http://www.geneious.com/). In addition, all genes were annotated with MITOS Web Server (http://mitos.bioinf.uni-leipzig.de/index.py) (Bernt et al. Citation2013) and tRNA scan-SE server (Lowe and Chan Citation2016).
The mitogenome of E. serrifemora was 14,947 bp, containing 22 transfer RNA genes (tRNAs), 13 protein-coding genes (PCGs), 2 ribosomal RNA genes (rrnL and rrnS). The region that we failed to sequence was between rrnS and trnI, and generally contained a putative AT-rich region. The composition of the genome contained 45.8% A, 15.7% C, 9.6% G, and 28.9% T, showing an obvious A + T bias (74.7%). Nine PCGs and 14 tRNA genes were transcribed from the majority strand, while the remaining four PCGs (ND1, ND4, ND4L, and ND5), eight tRNAs and two rRNAs were located on the minority strand. In addition, the gene composition and order were similar to all reported tetrigid species. Twelve PCGs started with typical ATN codon (seven with ATG, two with ATT, one with ATG and one with ATA), whereas the ND6 gene appeared to start with TTG. Eleven PCGs ended with complete stop codons (three with TAG, eight with TAA), and COIII and ND5 ended with the incomplete stop codons T (TA), which were presumably completed as TAA by post transcriptional polyadenylation (Anderson et al. Citation1981).
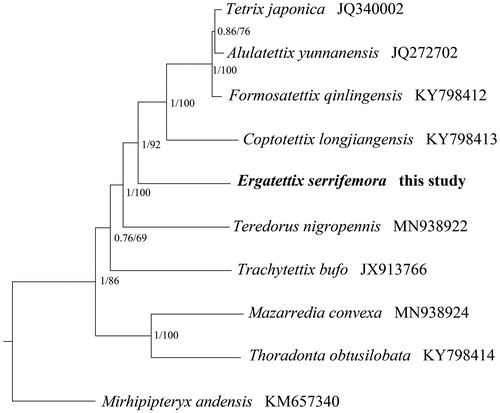
The phylogenetic relationships of E. serrifemora were reconstructed using Bayesian Inference (BI) by MrBayes 3.1.2 (Huelsenbeck and Ronquist Citation2001) and Maximum Likelihood (ML) by RAxML 8.2.0 (Stamatakis Citation2014), based on 13 PCGs (10,950 bp) from mitogenomes of night tetrigid species and one outgroup (Mirhipipteryx andensis), respectively. Two phylogenetic trees using different methods yielded the same topology, and nodal supporting values were always higher for BI tree than for ML tree (). The phylogenetic tree confirmed that E. serrifemora was closely related to other five species from Tetriginae, and all the species of the subfamily Tetriginae formed a clade. Presently, the studies recorded for Tetrigoidea was limited and we hope that our data can be useful for further study.
Figure 1. Phylogenetic tree obtained from ML and BI analysis based on 13 concatenated mitochondrial PCGs. Numbers on node are posterior probability (PP) and bootstrap value (BV).
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The data that support the findings of this study are openly available in [National Center for Biotechnology Information] at [https://www.ncbi.nlm.nih.gov/nuccore], reference number [MN938923].
Additional information
Funding
References
- Anderson S, Bankier AT, Barrell BG, de Bruijn MH, Coulson AR, Drouin J, Eperon IC, Nierlich DP, Roe BA, Sanger F, et al. 1981. Sequence and organization of the human mitochondrial genome. Nature. 290(5806):457–465.
- Bernt M, Donath A, Jühling F, Externbrink F, Florentz C, Fritzsch G, Pütz J, Middendorf M, Stadler PF. 2013. MITOS: improved de novo metazoan mitochondrial genome annotation. Mol Phylogenet Evol. 69(2):313–319.
- Deng WA. 2016. Taxonomic study of Tetrigoidea from China [Ph.D. dissertation]. Wuhan: Huazhong Agricultural University.
- Huelsenbeck JP, Ronquist F. 2001. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics. 17(8):754–755.
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, et al. 2012. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 28(12):1647–1649.
- Lowe TM, Chan PP. 2016. tRNAscan-SE On-line: integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 44(W1):W54–W57.
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30(9):1312–1313.