
Abstract
Water voles (Microtus richardsoni) are sensitive species distributed in the mountains of Canada (Alberta, British Columbia), and the United States of America (Idaho, Montana, Oregon, Utah, Washington, and Wyoming). We assembled the complete circular M. richardsoni mitogenome, which is 16,285 bp in length and encodes 13 protein-coding genes, 22 tRNA genes, and two rRNA genes. We estimated the phylogenetic tree of M. richardsoni and 24 related arvicoline species with two outgroup species: Phodopus roborovskii and Cricetus cricetus.
Water voles (Microtus richardsoni) are considered sensitive species by the USDA Forest Service because of their small populations and specific habitat requirements. They are highly dependent on freshwater streams in the mountains of Canada (Alberta, British Columbia), and United States of America (Idaho, Montana, Oregon, Utah, Washington, and Wyoming) (Klaus et al. Citation2001).
We assembled the complete mitochondrial genome of a single M. richardsoni individual sampled from Hoodoo Lake, ID (46.333333 N, 114.633333 W) sequenced by Iridian Genomes, Inc. (Bethesda, MD; NCBI-SRA: accession number SRR8451905 and SRR8293759). Paired-end (2 × 150 bp) whole-genome sequencing (WGS) on an Illumina HiSeq X-Ten produced 186,970,325 and 117,371,326 read pairs, respectively. The percentage of mitochondrial read pairs in these libraries was estimated to be 0.91 and 1.63%, respectively. Moreover, the original specimen is in the Conner Museum at Washington State University, catalog number 04-1452.
We generated the mitogenome of M. richardsoni by using the SMART2 pipeline (Alqahtani and Mandoiu Citation2020). SMART2 was used to jointly assemble the two WGS libraries, additionally using a M. richardsoni cytochrome b gene sequence (GenBank ID AY973809) as seed sequence. The first step in SMART2 is automatic adapter detection and trimming. Second, SMART2 uses a doubling algorithm to automatically select the number of read pairs that are used to assemble the mitogenome. For M. richardsoni, the doubling algorithm selected 100,000 read pairs from each of the two WGS libraries. Next, SMART2 randomly resamples the selected number of trimmed read pairs from each library separately and filters the reads using a coverage-based filter based on the count distribution of the k-mers in the seed sequence. The fourth step is the preliminary assembly of reads passing the coverage-based filter using SPAdes (Bankevich et al. Citation2012). Fifth, preliminary contigs are filtered using BLAST (BLAST: Basic Local Alignment Search Tool Citation2019) searches against a local mitochondrial database. The sixth step is secondary filtering of the reads in the two libraries by alignment to preliminary contigs that have significant BLAST matches. The seventh step is a secondary de novo assembly using SPAdes for reads passing the alignment filter. Next, iterative scaffolding and gap filling are performed based on maximum likelihood. Lastly, mitochondrial genes are predicted and annotated using the MITOS annotation pipeline (Bernt et al. Citation2013). Steps 2-8 were repeated five times on randomly resampled sets of reads, generating an identical mitogenome sequence in all five runs (5/5 bootstrap support). Across both WGS libraries, the average number of mitochondrial reads assembled in each bootstrap sample was 3,527.8 ± 84.5884, with an average coverage is of 65×.
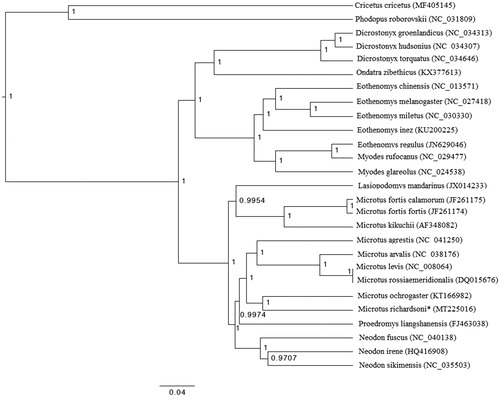
The mitogenome sequence (GenBank ID MT225016) generated by SMART2 has a length of 16,285bp. The mitogenome sequence annotation generated by MITOS has 13 protein-coding genes, 22 transfer RNA genes, and 2 ribosomal RNA genes. For our phylogenetic analysis, we downloaded 24 publicly available arvicoline mitogenomes and two outgroup sequences from GenBank and aligned them with our M. richardsoni mitogenome using the local pair algorithm in MAFFT (Katoh et al. Citation2002). The phylogeny was estimated using BEAST 2.5.0 (Bouckaert et al. Citation2019) with 10,000,000 Markov steps, 100,000 burn-in steps, and a GTR + I+G substitution model selected using jmodeltest (Posada Citation2008). The tree was visualized with FigTree (Rambaut and Drummond Citation2012). The phylogeny () shows M. richardsoni as sister to M. ochrogaster and paraphyly within the genus Microtus.
Acknowledgements
We would like to thank Jeffrey Good for collecting this tissue sample of M. richardsoni.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability
Raw data is available in NCBI at https://www.ncbi.nlm.nih.gov/sra/ with accession numbers are SRR8451905 and SRR8293759. Also, the mitogenome sequence of Microtus Richardsoni is available in NCBI at https://www.ncbi.nlm.nih.gov/nuccore with accession number MT225016.
Figure 1. Phylogenetic tree based on the complete mitochondrial genomes of 25 arvicoline species and two outgroups (Cricetus cricetus and Phodopus roborovski). The scale bar represents the number of substitutions per site and node labels represent posterior probabilities.
References
- Alqahtani F and Mandoiu II. 2020. SMART2: multi-library statistical mitogenome assembly with repeats. In: Mandoiu I, Murali T, Narasimhan G, Rajasekaran S, Skums S, and Zelikovsky A (eds). Computational advances in bio and medical sciences. Lecture Notes in Bioinformatics. Vol. 12029. Springer, Cham.
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, et al. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 19(5):455–477.
- National Center for Biotechnology Information, 2019. BLAST: Basic Local Alignment Search Tool.[Accessed July 1, 2019]. https://blast.ncbi.nlm.nih.gov/.
- Bernt M, Donath A, Jühling F, Externbrink F, Florentz C, Fritzsch G, Pütz J, Middendorf M, Stadler PF. 2013. MITOS: improved de novo metazoan mitochondrial genome annotation. Mol Phylogenet Evol. 69(2):313–319.
- Bouckaert R, Vaughan TG, Barido-Sottani J, Duchener S, Fourment M, Gavryushkina A, Heled J, Jones G, Kuhnert D, Maio ND, et al. 2019. BEAST 2.5: an advanced software platform for Bayesian evolutionary analysis. PLOS Comput Biol. 15(4):e1006650.
- Katoh K, Misawa K, Kuma K-i, Miyata T. 2002. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30(14):3059–3066.
- Klaus M, Moore RE, Vyse E. 2001. Microgeographic variation in allozymes and mitochondrial DNA of Microtus richardsoni, the water vole, in the Beartooth Mountains of Montana and Wyoming, USA. Canadian J Zool. 79(7):1286–1295.
- Posada D. 2008. jModelTest: phylogenetic model averaging. Mol Biol Evol. 25(7):1253–1256.
- Rambaut A, Drummond AJ. 2012. FigTree version 1.4.0. [Accessed July 1, 2019]. https://github.com/rambaut/figtree.