
Abstract
The complete chloroplast genome of Stachys japonica was reconstructed by reference-based assembly using Illumina paired-end data. The assembled plastome is 150,599 base pairs (bp) in length, including a pair of inverted repeat regions (IRs) of 25,654 bp each, a large single-copy region (LSC) of 81,701 bp and a small single-copy region (SSC) of 17,590 bp. A total of 131 genes were predicted from the chloroplast genome, including 86 protein coding genes, 37 tRNA genes and 8 rRNA genes. The overall GC content of S. japonica chloroplast genome was 38.5%. Phylogenetic analysis with several reported chloroplast genomes showed that S. japonica is closely clustered with S. sylvatica. The complete chloroplast genome of S. japonica provides new insight into Labiatae evolutionary and genomic studies.
The perennial herb Stachys japonica is widely distributed in China, Japan and Russia. It has been used in traditional Chinese medicine for the treatment of dysentery, laryngitis and a few other diseases. To date, the phylogenetic position of S. japonica in the genus Stachys is still unclear. In this study, we first reported the complete chloroplast (cp) genome of S. japonica and reconstructed a plastome phylogeny for the genus Stachys.
The mature and healthy leaves of a single individual of S. japonica was sampled from the field of Haiyuan county in south Ningxia, NW China (36°12′43″N, 105°37′9″E). The voucher specimen was deposited in the Herbarium of Sichuan University (accession number: QTPLJQ14383116). The total genomic DNA was extracted from silica gel dried leaves using a modified CTAB method (Doyle and Doyle Citation1987) and sequenced based on the Illumina pair-end technology. The filtered reads were assembled using the program NOVOPlasty (Dierckxsens et al. Citation2017) with complete cp genome of S. coccinea as the reference (GenBank accession no. NC_029823). The assembled cp genome was annotated using Plann (Huang and Cronk Citation2015), and the annotation was corrected using Geneious (Kearse et al. Citation2012). To examine the phylogenetic position of S. japonica, a multiple sequence alignment (MSA) analyses was performed using MAFFT v7.313 (Katoh and Standley Citation2013) based on seven cp genomes in Labiatae. Finally, a maximum likelihood (ML) tree was constructed by RAxML v8.2.11 (Stamatakis Citation2006) with 500 bootstrap replicates based on the alignments, using two species from the genus Lamium as outgroup.
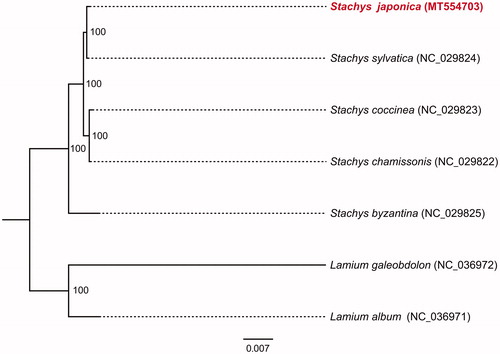
The complete cp genome of S. japonica was a circular molecular genome with a size of 150,599 bp in length, which presented a typical quadripartite structure containing two inverted repeat (IR) regions of 25,654 bp separated by the large single-copy (LSC) region of 81,701 bp and small single-copy (SSC) region of 17,590 bp. The cp genome consists of 131 genes including 86 protein coding genes, 37 tRNA genes, and 8 rRNA genes. The overall GC content was about 38.5%. In the plastome phylogeny, S. japonica shows the closest genetic relationship to S. sylvatica. (100% bootstrap support) (). The S. japonica cp genome can be further used for population genomic studies, phylogenetic analyses, genetic engineering studies of Labiatae.
Figure 1. Phylogenetic relationships of seven species based on chloroplast genome sequences. Bootstrap support is indicated for each branch..
Acknowledgements
The authors thank Dr. Lei Zhang from Sichuan University for the help of plant material collection.
Disclosure statement
None of the coauthors has any conflict of interest to declare. The authors alone are responsible for the content and writing of the paper.
Data availability statement
The data that support the findings of this study are openly available in NCBI GenBank database at https://www.ncbi.nlm.nih.gov/ with the accession number is MT554703 or available in [figshare.com] at https://doi.org/10.6084/m9.figshare.12443378.
Additional information
Funding
References
- Dierckxsens N, Mardulyn P, Smits G. 2017. NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 45(4):e18.
- Doyle JJ, Doyle JL. 1987. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull. 19:11–15.
- Huang DI, Cronk Q. 2015. Plann: a command-line application for annotating plastome sequences. Appl Plant Sci. 3(8):1500026.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30(4):772–780.
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, et al. 2012. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 28(12):1647–1649.
- Stamatakis A. 2006. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 22(21):2688–2690.