
Abstract
Sycopsis sinensis is a species of the genus Sycopsis Oliv in the Hamamelidaceae, and it is a native broadleaved evergreen woody plant species in China. Here, we sequenced, assembled, and analyzed the complete chloroplast (cp) genome of S. sinensis. The chloroplast genome of S. sinensis was 159,093 bp with 38.02% GC content, including a large single-copy (LSC) region of 87,841 bp, a small single-copy (SSC) region of 18,792 bp and two equal length inverted repeat (IR) regions of 26,230 bp. And, it contained 131 genes, including 37 tRNA genes, 8 rRNA genes, and 86 mRNA genes. Phylogenetic analysis strongly shows that S. sinensis has a close relationship with Distylium macrophyllum, whose posterior probability is 1.0.
Sycopsis sinensis Oliver, belonging to genus Sycopsis in Hamamelidaceae(Li et al., 2003), is a native broadleaved evergreen tree species distributed in Southen China. The largest population of S. sinensis as the dominant species was reported in the Houhe National Nature Reserve (Liu et al. Citation1999). Ago, domestic research on S. sinensis was limited to analysis of flora components and community level, but the research on S. sinensis from molecule level has not been reported so far. Here we firstly reported the complete chloroplast genomes of S. sinensis to promote further studies on population genetics, phylogeny and conservation biology in Hamamelidaceae.
Fresh leaves from one individual tree of S. sinensis were collected at the campus of Nanjing Forestry University (118.809603°E, 32.078520°N). The voucher specimen has been preserved at the Herbarium of Nanjing Forestry University (accession number Duan19121401). The whole genome was sequenced using Illumina NovaSeq highthroughput Sequencing platform based on Sequencing By Synthesis (SBS) technology. As a result, total 22,748,960 clean reads were produced and then used for the de novo assembly with NOVOplasty 2.7.2 (Dierckxsens et al. Citation2016). The annotated sequence has been submitted to GenBank with the accession number MT323104. Genome annotation, visualization, and tandem repeats identificationwas performed using the CpGAVAS pipeline (Liu et al. Citation2012).
The complete chloroplast genomes of S. sinensis was 159,093 bp. Like other species' chloroplast genome of Hamamelidaceae, the genome had a pair of IRs (26,230bp) regions separated by the LSC (87,841bp) and SSC (18,792bp) regions. The overall GC content was 38.02% for S. sinensis, whereas the GC content in the LSC, SSC and IR regions were36.19%, 32.47% and 43.06%, respectively. It contained 131 genes, including 37 tRNA genes, 8 rRNA genes, and 86 mRNA genes. Among those, 112 were unique and 19 (ndhB, rpl2, rpl23, rps12, rps7, rrn16, rrn23, rrn4.5, rrn5, trnAUGC, trnI-CAU, trnI-GAU, trnL-CAA, trnN-GUU, trnR-ACG, trnV-GAC, ycf1, ycf15, and ycf2) were duplicated in IR regions. Fourteen genes (eight protein-coding genes and six tRNA genes) contained one intron, and three genes (clpP, rps12, and ycf3) contained two introns.
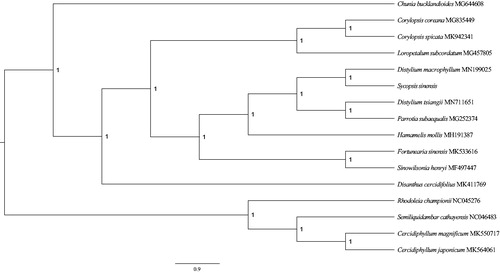
In order to show position of S. sinensis in Hamamelidaceae, a phylogenetic tree was conducted by using MrBayes 3.2.6 (Ronquist et al. Citation2012) to infer. We selected and downloaded 16 species’ sequences of their complete chloroplast genomes from the National Center for Biotechnology Information. Sequences was aligned with MAFFT (Rozewicki et al. Citation2019) using '–auto' strategy and normal alignment mode across PhyloSuite1.1.16 (Zhang et al. Citation2020). ModelFinder (Kalyaanamoorthy et al. Citation2017) was used to select the best-fit model (GTR + F + I + G4) using AIC criterion. A Markov chain Monte Carlo (MCMC) was run for 1,000,000 generations with two parallel searches using four chains, each starting with a random tree. Trees were sampled every 100 generations. From the phylogenetic tree, we could discover that all species were clustered on the correct clades of family, S. sinensis and its congeneric species, Distylium macrophyllum, as sister group (posterior probability = 1.0; ).
Figure 1. Phylogenetic relationships among 16 complete chloroplast genomes of Hamamelidaceae and Cercidiphyllaceae. Posterior probability values are given at the nodes. Cercidiphyllaceae are used as outgroups.
Acknowledgments
We would like to thank Doctor Yao Li from Nanjing Forestry University for his help with fieldwork; Lei Xie and Lu Wang from Nanjing Forestry University for their assistant with data analyses.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Dierckxsens N, Mardulyn P, Smits G. 2016. NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 45(18):e18.
- Kalyaanamoorthy S, Minh BQ, Wong TKF, von Haeseler A, Jermiin LS. 2017. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods. 14(6):587–589.
- Li X, Hu LL, Huang HD, Jiang MX. 2003. Structures and spatial distribution patterns of dominant populations in Sycopsis sinensis community in Houhe Nature Reserve. J. App1. Ecol. 14(6):849–852.
- Liu C, Shi L, Zhu Y, Chen H, Zhang J, Lin X, Guan X. 2012. CpGAVAS, an integrated web server for the annotation, visualization, analysis, and GenBank submission of completely sequenced chloroplast genome sequences. BMC Genomics. 13:715.
- Liu SX, Xu KY, Yang F-S. 1999. The biggest community Sycopsis sinensis was found in Houhe Nature Reserve, Hubei Province, China. J Huazhong nor Univ (Nat Sci). 33(4):588–589.
- Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Hohna S, €Larget B, Liu L, Suchard MA, Huelsenbeck JP. 2012. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 61(3):539–542.
- Rozewicki J, Li S, Amada KM, Standley DM, Katoh K. 2019. MAFFT-DASH: integrated protein sequence and structural alignment. Nucleic Acids Res. 47(W1):W5–W10.
- Zhang D, Gao F, Jakovlić I, Zou H, Zhang J, Li WX, Wang GT. 2020. PhyloSuite: an integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol Ecol Resour. 20(1):348–355.