
Abstract
In this study, we obtained the complete mitochondrial genome of Hapalogenys analis using whole genome sequencing. With the exception for control region, this mitochondrial genome, consisting of 16,355 base pairs (bp), contains 13 protein-coding genes (PCGs), 2 ribosomal RNAs (rRNAs), and 21 transfer RNAs (tRNAs). This mitochondrial genome also lacks a tRNA-Pro gene after tRNA-Thr gene. The overall base composition shows 25.45% of T, 29.73% of C, 28.68% of A and 16.14% of G, with a slight A + T rich feature (54.13%). Sanger sequencing is needed to confirm the accuracy of control region, as well as the lack of tRNA-Pro gene. The mitogenome data provides useful genetic markers for the studies on the molecular identification, population genetics, phylogenetic analysis and conservation genetics.
The Hapalogenys anails is an important economic species distributed mainly in the northwestern Pacific, including the coastal waters of southern China and Japan (Xu et al. Citation2010). This species is a kind of demersal fish living in the water depth of 30–50 m, and often occurs in artificial reef areas (Xu et al. Citation2010). To date, the taxonomic status of genus Hapalogenys is controversial and problematic (Wei et al. Citation2014). However, only limited genetic information have been published for H. anails. In this study, the next-generation sequencing technology is used to determine the complete mitochondrial genome of H. anails. The structure of mitogenome is compared and analyzed, and some gene markers are further used for the phylogenetic status of this species.
The sample of H. anails was collected from the Xixuan Island coastal waters of Zhoushan (29.54°N, 122.19°E), China in September 2019. The examined specimen was preserved at Fisheries Ecology and Biodiversity Laboratory in Zhejiang Ocean University under specimen accession no. ZJOU-03742. The genomic DNA was extracted from dorsal-lateral muscles (30 mg) using Rapid Animal Genomic DNA Isolation Kit (Sangon Biotech Co., Ltd., Shanghai, CN). A genomic library was established and followed by the next-generation sequencing. Whole genome sequencing (sequencing depth 50X) was conducted using Illumina Hiseq4000 platform with the sequencing insertion of 350-bp. Quality check for sequencing data was done by FastQC (Andrews Citation2010) and the filtered clean data were assembled and mapped to complete mitogenome sequence using NOVOPlasty v3.7.2 (Dierckxsens et al. Citation2017). Subsequently, the assembled sequence was annotated using the online Mitochondrial Genome Database of Fish server (Iwasaki et al. Citation2013) and the MITOS Web Server (Bernt et al. Citation2013).
The complete mitogenome was assembled using NOVOPlasty software initially. However, with the lack of tRNA-Pro, we doubted the accuracy of control region sequences. As a result, the final sequence except for tRNA-Pro and control region has been deposited in GenBank with accession number MT561534. The mitochondrial genome of H. anails in this study (16,355 bp in length) consists of 13 protein-coding genes, 21 tRNA genes, and 2 rRNA genes. The arrangement of all genes is identical to those of most vertebrates (Wang et al. Citation2008; Chen Citation2013; Chiang et al. Citation2013). Most of the genes are encoded on the heavy strand (H-strand), except for the eight tRNA genes (-Gln, -Ala, -Asn, -Cys,-Tyr, -Ser, and -Glu) and one protein-coding gene (ND6). The overall base composition is 25.45% for T, 29.73% for C, 28.68% for A, and 16.14% for G, with a slight A + T-rich feature (54.13%). Except for COI starting with GTG, the remaining 12 protein-coding genes start with ATG. It is important to note that some of the protein-coding genes (6 of 13 genes) are inferred to terminate with an incomplete stop codon (COII, COIII, ND2, ND3, ND4 and Cyt b), with six (COI, ATPase8, ATPase6, ND1, ND4L and ND5) sharing TAA and one (ND6) using TAG as a stop codon, respectively. These features are common among vertebrate mitochondrial genome, and TAA is supposed to be appeared via posttranscriptional polyadenylation (Ojala et al. Citation1981). The longest one is ND5 gene (1842 bp) among protein-coding genes, whereas the shortest is ATPase 8 gene (168 bp). The two ribosomal RNA genes, 12S rRNA (953 bp) and 16S rRNA (1697 bp), are located between tRNAPhe and tRNALeu.
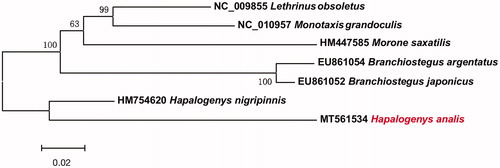
Phylogenetic relationships were constructed using neighbour-joining (NJ) algorithm implemented in MEGA 6 (Tamura et al. Citation2013) among 6 species of family Haemulidea based on 12 H-strand mitochondrial protein-coding genes (). This phylogenetic tree showed that H. anails has a relatively close relationship with H. nigripinnis. The information of the mitogenome will be useful for future phylogenetic studies and specimen identification of Haemulidea species.
Figure 1. Neighbor-joining (NJ) topology for 6 species of family Haemulidae based on 12 H-strand mitochondrial protein-coding genes.
Acknowledgement
We are grateful to Ms. Yuan Zhang and Xiaoyan Wang for the help of sampling.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The data that support the findings of this study is openly available in GenBank of NCBI at https://www.ncbi.nlm.nih.gov under accession number MT561534.
Additional information
Funding
References
- Andrews S. 2010. FastQC: a quality control tool for high throughput sequence data [cited 2018 Oct 4]. Available from: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
- Bernt M, Donath A, Jühling F, Externbrink F, Florentz C, Fritzsch G, Pütz J, Middendorf M, Stadler PF. 2013. MITOS: improved de novo metazoan mitochondrial genome annotation. Mol Phylogenet Evol. 69(2):313–319.
- Chen IS. 2013. The complete mitochondrial genome of Chinese sucker Myxocyprinus asiaticus (Cypriniformes, Catostomidae). Mitochondrial DNA. 24(6):680–682.
- Chiang TY, Chen IS, Lin HD, Chang WB, Ju YM. 2013. Complete mitochondrial genome of Sicyopterus japonicus (Perciformes, Gobiidae). Mitochondrial DNA. 24(3):191–193.
- Dierckxsens N, Mardulyn P, Smits G. 2017. NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 45(4):e18–e18.
- Iwasaki W, Fukunaga T, Isagozawa R, Yamada K, Maeda Y, Satoh TP, Sado T, Mabuchi K, Takeshima H, Miya M, et al. 2013. MitoFish and MitoAnnotator: a mitochondrial genome database of fish with an accurate and automatic annotation pipeline. Mol Biol Evol. 30(11):2531–2540.
- Ojala D, Montoya J, Attardi G. 1981. tRNA punctuation model of RNA processing in human mitochondria. Nature. 290(5806):470–474.
- Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. 2013. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 30(12):2725–2729.
- Wang CH, Chen Q, Lu GQ, Xu JW, Yang QL, Li SF. 2008. Complete mitochondrial genome of the grass carp (Teleostei, Cyprinidae, Gobioninae). Gene. 424(1–2):96–101.
- Wei T, Sun YN, Zhang B, Wang RX, Xu TJ. 2014. A mitogenomic perspective on the phylogenetic position of the Hapalogenys genus (Acanthopterygii: Perciformes) and the evolutionary origin of Perciformes. PLoS One. 9(7):e103011
- Xu TJ, Wang JX, Sun YN, Shi G, Wang RX. 2010. Phylogeny of Hapalogenys with discussion on its systematic position in Precoidea using Cytochrome b gene sequences. Acta Zootaxon Sin. 35:530–536 (In Chinese with English abstract).