
Abstract
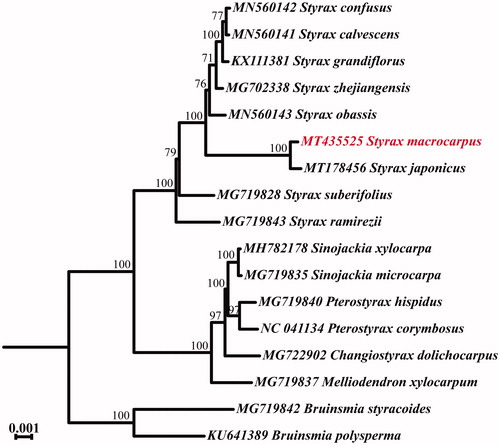
The whole chloroplast genome of Styrax macrocarpus, an endemic species distributed in China, is determined in this study. The whole chloroplast genome size is 157,805 bp in length, containing a large single-copy region (LSC, 87,628 bp) and a small single-copy region (SSC, 18,267 bp), which were separated by a pair of inverted repeats (IRs, 25,955 bp). One hundred thirty one genes were detected from this genome, including 85 protein-coding genes, 38 transfer RNA genes, and 8 ribosomal RNA genes. The phylogenetic analysis suggested that the S. macrocarpus is closely clustered with S. japonicus with strong support values.
Styrax macrocarpus Cheng (Styraceae) is an endemic species that is distributed in Hunan and Guangdong provinces of China with elevations of 500–850 m (Cheng Citation1987). Recently, the original habitat of S. macrocarpus is suffering from severe damage from human activities and it is already critically endangered and more conservation strategies should be implemented urgently (Peng et al. Citation2011). However, no research has been done on this endangered species so far except a phylogenetic and biogeographical study on the genus Styrax (Fritsch Citation2001). In this study, we assembled and characterized the complete chloroplast genome of S. macrocarpus (the GenBank accession number is MT435525) using the Illumina paired-end sequencing data.
Fresh leaves were collected from Yizhang county, Hunan province, China (coordinates: 24°56′48.39″N, 112°59′00.68″E) and the voucher specimens were deposited in the Herbarium of Changsha Environmental Protection College (accession number is DG-2019-002). Total genomic DNA was extracted from leaf tissue with modified CTAB method (Doyle and Doyle Citation1987), and the whole-genome sequencing was conducted on the Illumina Hiseq Platform (Novogene, Beijing, China). After quality test, the complete chloroplast genome was assembled via NOVOPlasty program (Dierckxsens et al. Citation2017) with the cp genomes of Styrax suberifolius (GenBank accession number is MG719828) as the best reference. The cp genome was further annotated using software Geneious v11.0.3 (Kearse et al. Citation2012) and the genome map was generated using the web server OGDRAW (http://ogdraw.mpimp-golm.mpg.de/) (Lohse et al. Citation2013).
The complete cp genome of S. macrocarpus was a double-stranded circular DNA with 157,805 bp in size that compose of a large single copy (LSC, 87,628 bp), a small single copy (SSC, 18,267 bp), and a pair of inverted repeat regions (IRs, 25,955 bp). One hundred thirty one genes were detected, containing 85 protein-coding genes, 38 transfer RNA genes, and 8 ribosomal RNA genes, among them, there are 17 duplicated genes, including 6 protein-coding genes, 4 ribosomal RNA genes, and 7 transfer RNA genes. The overall GC content of S. macrocarpus complete cp genome is 37.0%.
For phylogenetic analysis, 16 cp genome sequences from genus Styrax and related species in family Styracaceae were downloaded from National Center for Biotechnology Information (NCBI, https://www.ncbi.nlm.nih.gov/). All cp genomes were aligned using MAFFT (Katoh et al. Citation2002) and then used to construct the maximum likelihood (ML) tree via RAxML v8 (Stamatakis Citation2014) with 1000 bootstrap replicates (). The result revealed that S. japonicus has close relationship with S. japonicus. The cp genome of S. macrocarpus will provide important information for the conservation of this species.
Figure 1. Phylogenetic relationships between the S. macrocarpus and other 16 complete chloroplast genome sequences from family Styracaceae. The bootstrap support values are shown on the branches. Position of the S. macrocarpus was labeled with red font.
Disclosure statement
The authors are grateful to the opened raw genome data from public database. No potential conflict of interest was reported by the author(s).
Data availability statement
The sequence of Styrax macrocarpus used in this article is available at the NCBI database with accession number: MT435525 (https://www.ncbi.nlm.nih.gov/nuccore/MT435525).
References
- Cheng WC. 1987. Flora of China. Vol. 60. Beijing: Science Press; P. 97–98.
- Dierckxsens N, Mardulyn P, Smits G. 2017. NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 45(4):e18.
- Doyle JJ, Doyle JL. 1987. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull. 19:11–15.
- Fritsch PW. 2001. Phylogeny and biogeography of the flowering plant genus Styrax (Styracaceae) based on chloroplast DNA restriction sites and DNA sequences of the internal transcribed spacer region. Mol Phylogenet Evol. 19(3):387–408.
- Katoh K, Misawa K, Kuma K, Miyata T. 2002. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30(14):3059–3066.
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, et al. 2012. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 28(12):1647–1649.
- Lohse M, Drechsel O, Kahlau S, Bock R. 2013. Organellar Genome DRAW–a suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 41:575–581.
- Peng ZH, Zhang C, Wu T, Peng Y. 2011. Resource of the Styracaceae in Hunan and their application to landscaping. Guangdong Agr Sci. 7:76–78.
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30(9):1312–1313.