
Abstract
Species of the Sternoptychidae teleost family display an impressive morphology, including their extreme reduced size. Here, we report the first mitochondrial genome of the diaphanous hatchet fish Sternoptyx diaphana. By using short-read sequencing Illumina HiSeq, we generated two mitochondrial contigs which were later physically assembled by PCR. The mitochondrial genome of S. diaphana was 17,224 bp in length (excluding the control region) and is composed of 13 PCGs and 2 ribosomal RNA genes. Strikingly, we could not identify the tRNA-Phe and two copies of tRNA-Met were differently positioned. Additionally, the mitogenome displays a completely new gene rearrangement among vertebrates. We expect that the study presented here will pave the way for further molecular studies with this underrepresented group of illusive teleost fish.
Members of the marine teleost Sternoptychidae family (Stomiiformes) are characterized by their extremely small size (<100 mm). Moreover, they display a bright silver lateral pigmentation, large eyes, numerous lateral and ventral photophores, and a highly variable body morphology which is gender-specific (Nelson et al. Citation2016). The family consists of two subfamilies, the Maurolicinae and Sternoptychinae (Weitzman Citation1974), which include 70 species distributed over 10 genera. One of these species Sternoptyx diaphana Hermann 1781, is a very small (up to 45 mm) deep-sea ray-finned fish, with large eyes and short snout, and a laterally compressed body, with the mouth being nearly vertical.
A specimen of S. diaphana was captured in North Atlantic (48.0695 N; 47.0857 W) from the EU Groundfish Survey (Fletán Negro 3 L-2019) and stored in ethanol 96%. Due to its small dimension, the whole individual (except the digestive tract) was used for total genomic DNA extraction and subsequent whole-genome library preparation (350 bp Truseq DNA PCR-free Illumina kit) and sequencing (150 bp Paired-end on HiseqX150) done at Macrogen Inc., Korea. Nevertheless, other specimens of the same species simultaneously collected are stored in ethanol at Interdisciplinary Center of Marine and Environmental Research and the Instituto Español de Oceanografía, Centro Oceanográfico de Vigo with vouchers S.DIAPNA_1_3L19 and S.DIAPNA_2_3L19, respectively.
Mitogenome assembly was performed using a subsample of the whole genome sequencing using mode all of MitoZ (Meng et al. Citation2019). Despite various attempts, it was not possible to obtain a single contig from the NGS data alone, with a final assembly being constituted of two contigs. To scaffold over the two contigs, two primers matching the edge of both contigs, P8F 5′-GGTATTTGGTGCTTGAGC-3′ P8R and 5′-GCTAACACAAATACCCAGTCCG-3′, were designed and used for PCR amplification and Sanger sequencing. Each PCR reaction contained 2.5 μL 10× Invitrogen PCR Buffer, 1.5 μL 50 mmolL−1 MgCl2, 0.5 μL 10 mmolL−1 of each primer, and 10 mmolL−1 dNTPs, 0.1 μL Invitrogen Taq DNA Polymerase and approximately 1 μL DNA template. The cycle conditions (repeated for 36×) were as follows, initial denaturation at 94 °C for 3 min, denaturation at 94 °C (30 s), annealing at 54 °C (40 s), and extension at 72 °C (60 s) with a final extension at 72 °C for 10 min. Chromatograms were visually inspected using ChromasPro version 1.41 (technelysium.com.au) and scaffolding performed manually using BioEdit version 5.0.9 (Hall Citation1999). The final mitogenome was annotated in MITOS2 (Bernt et al. Citation2013) and manually validated by comparison with annotations from other Stomiiformes available at GenBank.
For phylogenetic analysis, all Stomiiformes mitogenomes (n = 7) and five outgroup taxa, were retrieved from GenBank (accessed in May 2020). BI and ML phylogenetic analysis were performed using all 13 protein-coding genes (PCG), individually aligned using GUIDANCE2 (Sela et al. Citation2015) with MAFFT (Katoh and Standley Citation2013) and concatenated in SequenceMatrix (Vaidya et al. Citation2011). The final alignment was 11,349 nt long. PartitionFinder2 on XSEDE (Lanfear et al. Citation2016) was applied to determine the partition schemes and best molecular evolutionary models for those partitions and the retrieved information used for phylogenetic analyses. These analyses were conducted using MrBayes on XSEDE (Ronquist et al. Citation2012) (GTR + I+G, GTR + I+G, HKY + I+G, and HKY + G) with two independent runs (107 generations, sampling frequency one tree for 1000 generations) and RAxML-HPC BlackBox (Stamatakis, Citation2014) (GTR + I+G). Both phylogenetic analyses and PartitionFinder2 were implemented through CIPRES (Miller et al. Citation2010).
The mitogenome of S. diaphana here obtained represents the first mitogenome of a deep-sea fish from the family Sternoptychidae and has been deposited in GenBank (MT588184).
The mitogenome length (control region excluded) is 17,224 bp, within the observed size of other Stomiiformes mitogenomes (Miya et al. Citation2001; Miya and Nishida Citation1999, Citation2000; Aguilar et al. Citation2018; Ijichi et al. Citation2018). Regarding gene content, as expected 13 PCGs and 2 ribosomal RNA genes are present. Although 22 transfer RNAs were also annotated, we could not detect tRNA-Phe and two copies of tRNA-Met were differently positioned in the mitogenome. Furthermore, the S. diaphana mitogenome shows a completely new gene rearrangement among vertebrates (Satoh et al. Citation2016): -trnC -trnQ nad2 -trnY trnW cox1 trnD -trnS2 cox2 trnK atp8 atp6 cox3 trnG nad3 trnR nad4l nad4 trnH trnS1 trnL1 nad5 -trnE cob trnT nad1 trnI trnM trnL2 trnN trnA -nad6 -trnP -trnM rrnS trnV rrnL. Notwithstanding, despite several attempts, we could not circularize the mitogenome and the control region could not be detected. The inability to detect both the control region and the tRNA-Phe (generally adjacent to the control region) may result from the limitation of using Illumina short-read sequencing (Goodwin et al. Citation2016) and therefore requires future validation.
Both BI and ML phylogenetic trees, rooted with Coregonus lavaretus (Linnaeus, 1758), Salmonidae (following Ijichi et al. Citation2018), show the same topology (). The phylogenetic analysis separates with high support, the group Stomiiformes from a cluster formed by families Synodontidae, Ateleopodidae, Myctophidae, and Trachipteridae.
Figure 1. Bayesian Inference phylogenetic tree based on 13 concatenated protein-coding genes from 12 mitogenomes, including 7 Stomiiformes species and five outgroup taxa. Only supports above 92% are shown. The asterisk (*) on the nodes indicate that both posterior probabilities and bootstrap support values are 100%.
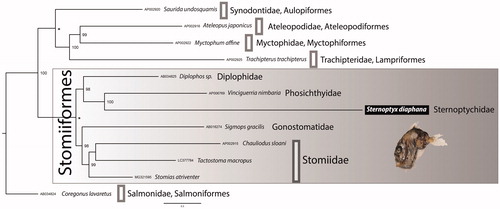
The Order Stomiiformes is divided into two well-supported groups, one comprising two families, i.e. Gonostomatidae and Stomiidae, and the other including Phosichthyidae, Diplophidae, and Sternoptychidae, represented by the here newly sequenced species S. diaphana. Although we have included all Stomiiformes available mitogenomes, this number is still reduced reinforcing the need to increase data sampling.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The data that supports the findings of this study are available in GenBank of NCBI at https://www.ncbi.nlm.nih.gov, reference number MT588184 or from the corresponding authors, Elsa Froufe and L. Filipe C. Castro.
Additional information
Funding
References
- Aguilar A, Truong BR, Gutierrez F. 2018. Complete mitochondrial DNA genomes for two northeast Pacific mesopelagic fishes, the Mexican lampfish (Triphoturus mexicanus) and black-belly dragonfish (Stomias atriventer). Mitochondrial DNA Part B. 3(1):21–23.
- Bernt M, Donath A, Jühling F, Externbrink F, Florentz C, Fritzsch G, Pütz J, Middendorf M, Stadler PF. 2013. MITOS: improved de novo metazoan mitochondrial genome annotation. Mol Phylogenet Evol. 69(2):313–319.
- Goodwin S, McPherson JD, McCombie WR. 2016. Coming of age: ten years of next-generation sequencing technologies. Nat Rev Genet. 17(6):333–351.
- Hall T. 1999. BioEdit: a user friendly biological sequence alignment editor and analysis program for windows 95/98/NT. Nuclear Acids Symposium Series, 41, 95–98.
- Ijichi M, Takano T, Hasegawa M, Yashiki H, Kogure K, Kojima S, Yoshizawa S. 2018. The complete mitochondrial genome of the longfin dragonfish Tactostoma macropus (Stomiiformes: Stomiidae). Mitochondrial DNA Part B. 3(2):486–487.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30(4):772–780.
- Lanfear R, Frandsen PB, Wright AM, Senfeld T, Calcott B. 2016. Partition finder 2: new methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol Biol E. 34:772–773.
- Meng G, Li Y, Yang C, Liu S. 2019. MitoZ: a toolkit for animal mitochondrial genome assembly, annotation and visualization. Nucleic Acids Res. 47(11):e63.
- Miller MA, Pfeiffer W, Schwartz T. 2010. Creating the CIPRES science gateway for inference of large phylogenetic trees. In: 2010 Gateway computing environments workshop, GCE 2010. Piscataway (NJ): IEEE; p. 1–8.
- Miya M, Kawaguchi A, Nishida M. 2001. Mitogenomic exploration of higher teleostean phylogenies: a case study for moderate-scale evolutionary genomics with 38 newly determined complete mitochondrial DNA sequences. Mol Biol Evol. 18(11):1993–2009.
- Miya M, Nishida M. 1999. Organization of the mitochondrial genome of a deep-sea fish, Gonostoma gracile (Teleostei: Stomiiformes): first example of transfer RNA gene rearrangements in bony fishes. Mar Biotechnol. 1(5):416–426.
- Miya M, Nishida M. 2000. Use of mitogenomic information in Teleostean molecular phylogenetics: a tree-based exploration under the maximum-parsimony optimality criterion. Mol Phylogenet Evol. 17(3):437–455.
- Nelson JS, Grande TC, Wilson MV. 2016. Fishes of the world. Hoboken (NJ): John Wiley & Sons.
- Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Höhna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP. 2012. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 61(3):539–542.
- Satoh TP, Miya M, Mabuchi K, Nishida M. 2016. Structure and variation of the mitochondrial genome of fishes. BMC Genomics. 17:719.
- Sela I, Ashkenazy H, Katoh K, Pupko T. 2015. GUIDANCE2: accurate detection of unreliable alignment regions accounting for the uncertainty of multiple parameters. Nucleic Acids Res. 43(W1):W7–W14.
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30(9):1312–1313.
- Vaidya G, Lohman DJ, Meier R. 2011. SequenceMatrix: concatenation software for the fast assembly of multi-gene datasets with character set and codon information. Cladistics. 27(2):171–180.
- Weitzman SH. 1974. Osteology and evolutionary relationships of the Sternoptychidae with a new classification of Stomiatoid families. Bull Am Museum Nat His. 153(3):328–478.