
Abstract
We completed chloroplast genome of Scapania ampliata Steph., presenting distinct morphological features including yellowish brown, one-celled gemmae, and decurrent dorsal lobe. It is 118,026 bp long and has four subregions: 80,850 bp of large single copy (LSC) and 19,436 bp of small single copy (SSC) regions are separated by 8,870 bp of inverted repeat (IR) regions including 130 genes (86 protein-coding genes, 8 ribosomal RNAs, and 36 transfer RNAs). The overall guanine cytosine (GC) content is 34.0% and those in the LSC, SSC, and IR regions are 31.9%, 31.0%, and 46.3%, respectively. Phylogenetic trees show S. ampliata is clustered with Scapania ciliata.
Genus Scapania (Dumort.) Dumort. is a large genus of leafy liverworts covering 121 accepted species (Söderström et al. Citation2016). Recent molecular phylogenetic studies of genus Scapania is often described as new to science and rearranged for section and genus (Heinrichs et al. Citation2012; Bakalin et al. Citation2019). Scapania ampliata Steph., the arctic-boreal species in Scapania genus, is distributed in the East Asia (Bakalin Citation2010; Choi et al. Citation2012). S. ampliata is distinct from the other Scapania species due to yellowish brown, one-celled gemmae, and decurrent dorsal lobe (Choi et al. Citation2012). To understand its phylogenetic position, we completed chloroplast genome sequences of S. ampliata.
The plants of S. ampliata collected in Taebaek city, Republic of Korea (Voucher in Jeonbuk National University Herbarium (JNU); S.S. Choi, CS-1910671a; 37.101486N, 128.917547E), was used for extracting DNA with DNeasy Plant Mini Kit (QIAGEN, Hilden, Germany). Genome sequencing was performed using NovaSeq6000 at Macrogen Inc., Korea. Chloroplast genome was completed by Velvet 1.2.10 (Zerbino and Birney Citation2008), SOAPGapCloser 1.12 (Zhao et al. Citation2011), BWA 0.7.17 (Li Citation2013), and SAMtools 1.9 (Li et al. Citation2009) under the environment of the Genome Information System (GeIS; http://geis.infoboss.co.kr/). Geneious R11 11.0.5 (Biomatters Ltd, Auckland, New Zealand) was used for annotation based on S. ciliata chloroplast genome (NC_043786; Yu et al. Citation2019).
The chloroplast genome of S. ampliata (GenBank accession is MT644123) is 118,026 bp long (guanine cytosine (GC) ratio is 34.0%) and has four subregions: 80,850 bp of large single copy (31.9%) and 19,436 bp of small single copy (31.0%) regions are separated by 8,870 bp of inverted repeat (IR; 46.3%). It contained 130 genes (86 protein-coding genes, eight ribosomal RNAs (rRNAs), and 36 transfer RNAs (tRNAs)); nine genes (four rRNAs and five tRNAs) are duplicated in IR regions. Gene order of S. ampliata chloroplast is identical to that of Scapania ciliata. Among the other Bryophyte chloroplast genomes, Marchantia (Takenaka et al. Citation2000; Kwon et al. Citation2019) and Aneura (Wickett et al. Citation2008; Myszczyński et al. Citation2017) genera contain more than one chloroplasts, displaying that Marchantia chloroplasts is conserved gene order but Aneura chloroplasts show structural variations.
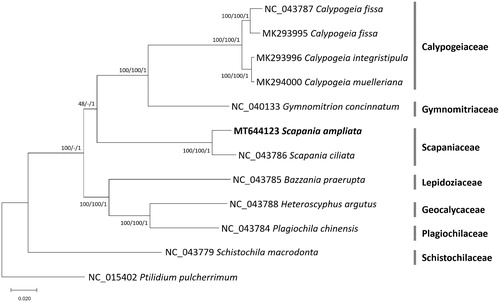
Eleven complete chloroplast genomes including S. ampliata were used for constructing neighbor joining (bootstrap repeat is 10,000), maximum likelihood (bootstrap repeat is 1,000), and Bayesian Inference (Number of generations is 1,100,000) phylogenic trees using MEGA X (Kumar et al. Citation2018) and Mr. Bayes (Huelsenbeck and Ronquist Citation2001) after aligning whole chloroplast genome sequences using MAFFT 7.450 (Katoh and Standley Citation2013). Phylogenetic trees show that two Scapania chloroplast genomes were clustered in one clade with high supportive values (). However, topology of the three clades, (Calypogeiaceae and Gymnomitriaceae), Scapaniaceae, and (Lepidoziaceae, Geocalycaceae, and Plagiochilaceae), is incongruent among the three phylogenetic trees (). Only neighbor joining tree displays different topology and the other two trees display high supportive values (); however, more investigation of inter-family phylogenetic relationship will be required with additional complete chloroplast genomes. Our chloroplast genome together with up-coming additional chloroplasts will provide better resolution of their phylogeny.
Figure 1. Neighbor joining (bootstrap repeat is 10,000), maximum likelihood (bootstrap repeat is 1,000), and Bayesian Inference phylogenetic trees (Number of generations is 1,100,000) of 12 complete chloroplast genomes: Scapania ampliata (MT644123 in this study), Scapania ciliata (NC_043786), Bazzania praerupta (NC_043785), Calypogeia fissa (NC_043787), Gymnomitrion concinnatum (NC_040133), Heteroscyphus argutus (NC_043788), Plagiochila chinensis (NC_043784), Schistochila macrodonta (NC_043779), Calypogeia fissa (MK293995), Calypogeia integristipula (MK293996), Calypogeia muelleriana (MK294000), and Ptilidium pulcherrimum (NC_015402) as an outgroup. Phylogenetic tree was drawn based on the maximum likelihood phylogenetic tree. Family names were displayed with grey bars in the right side of the tree. The numbers above branches indicate bootstrap support values of maximum likelihood, neighbor joining, and Bayesian Inference phylogenetic trees, respectively.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
Chloroplast genome sequence can be accessed via accession number MT644123 in NCBI GenBank.
Additional information
Funding
References
- Bakalin VA. 2010. Hepaticae of the Kuril Islands (northwestern Pacific): a transoceanic route from circumboreal to East Asian flora. Annales Botanici Fennici. 47(2):81–105.
- Bakalin V, Vilnet A, Ma WZ, Klimova K. 2019. The differentiation and speciation of Scapania javanica and S. undulata complexes in the Eastern Sino-Himalayas and perimeters for Scapania Sect. Stephania (Scapaniaceae, Hepaticae). Phytotaxa. Phytotaxa. 400(3):123–144.
- Choi SS, Bakalin VA, Sun B-Y. 2012. Scapania and Macrodiplophyllum in the Russian Far East. Bot Pac. 1 (1):31–95.
- Heinrichs J, Bombosch A, Feldberg K, Kreier H-P, Hentschel J, Eckstein J, Long D, Zhu R-L, Schäfer-Verwimp A, Schmidt AR, et al. 2012. A phylogeny of the northern temperate leafy liverwort genus Scapania (Scapaniaceae, Jungermanniales). Mol Phylogenet Evol. 62(3):973–985.
- Huelsenbeck JP, Ronquist F. 2001. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics. 17(8):754–755.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30(4):772–780.
- Kumar S, Stecher G, Li M, Knyaz C, Tamura K. 2018. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 35(6):1547–1549.
- Kwon W, Kim Y, Park J. 2019. The complete chloroplast genome of Korean Marchantia polymorpha subsp. ruderalis Bischl. & Boisselier: low genetic diversity between Korea and Japan. Mitochondrial DNA Part B. 4(1):959–960.
- Li H. 2013. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. [arXiv preprint] arXiv. :1303.3997.
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup. 2009. The sequence alignment/map format and SAMtools. Bioinformatics. 25(16):2078–2079.
- Myszczyński K, Bączkiewicz A, Buczkowska K, Ślipiko M, Szczecińska M, Sawicki J. 2017. The extraordinary variation of the organellar genomes of the Aneura pinguis revealed advanced cryptic speciation of the early land plants. Sci Rep. 7(1):9804.
- Söderström L, Hagborg A, Von Konrat M. 2016. Early land plants today: index of liverworts & hornworts 2013–2014. Phytotaxa. 269(3):133–185.
- Takenaka M, Yamaoka S, Hanajiri T, Shimizu-Ueda Y, Yamato KT, Fukuzawa H, Ohyama K. 2000. Direct transformation and plant regeneration of the haploid liverwort Marchantia polymorpha L. Transgenic Res. 9(3):179–185.
- Wickett NJ, Zhang Y, Hansen SK, Roper JM, Kuehl JV, Plock SA, Wolf PG, Depamphilis CW, Boore JL, Goffinet B. 2008. Functional gene losses occur with minimal size reduction in the plastid genome of the parasitic liverwort Aneura mirabilis. Mol Biol Evol. 25(2):393–401.
- Yu Y, Liu HM, Yang JB, Ma WZ, Pressel S, Wu YH, Schneider H. 2019. Exploring the plastid genome disparity of liverworts. J Syst Evol. 57(4):382–394.
- Zerbino DR, Birney E. 2008. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18(5):821–829.
- Zhao Q-Y, Wang Y, Kong Y-M, Luo D, Li X, Hao P. 2011. Optimizing de novo transcriptome assembly from short-read RNA-Seq data: a comparative study. BMC Bioinf. 12(Suppl 14):S2.