
Abstract
The complete chloroplast (cp) genome sequence of Pleurospermum amabile is characterized, and its phylogenetic relationships with related taxa in Apiaceae are revealed. The results showed that the complete cp genome of P. amabile was 155,955 bp in length, consisting of a large single-copy (LSC) region of 85,745bp, a small single-copy (SSC) region of 15,562 bp, which were separated by two inverted repeat regions (IRa and IRb) of 26,324bp, each. In total, 129 genes were annotated, comprising of 84 protein-coding genes, 37 tRNA genes, and 8 rRNA genes. The phylogenetic analysis indicated that P. amabile is a member of the East-Asia Clade, and showed a close relationship to Chuanminshen violaceum.
The genus Pleurospermum (Apiaceae), with about 50 species distributed in eastern Europe and north of Asia, is one of the difficult genera to delimit the circumscription and relationships with related taxa (Sheh et al. Citation2005; Valiejo-Roman et al. Citation2012). Phylogenomics is an effective approach to resolve relationships at various taxonomic levels, we therefore obtain the complete chloroplast (cp) genome sequence of this genus to get massive data for phylogenetic analysis. Pleurospermum amabile Craib & W. W. Smith is distributed in SE Xizang, NW Yunnan of China, and neighboring Bhutan, Sikkim (Sheh et al. Citation2005). It is distinguished by its membranous bracts and bracteoles with purple veins and has been used as a traditional medicine in Xizang.
Pleurospermum amabile was collected from Chayu (29°31′95.98″ N, 97°02′78.01′′ E), Xizang of China, and the voucher (Z15106) was deposited in Kunming Institute of Botany, Chinese Academy of Sciences (KUN). Total genomic DNA was extracted from leaf tissue using a Universal Genomic DNA Extraction kit (Tiangen Biotech, Beijing, China) following the manufacturer’s protocol. Then, the genome sequencing was performed with Illumina Hiseq 2500 (Majorbio, Shanghai, China) platform with the pair-end (2 × 300) library. The raw reads were filtered and assembled following the previously reported method (Zhou and Liu Citation2020) with Daucus carota (No. NC_008325) as the reference.
The cp genome of P. amabile exhibited a general quadripartite structure of 155,955 bp in length, with 37.7% over-all GC content. It contained a pair of IRs (inverted repeats) of 26,324 bp, separated by a small single-copy (SSC) region of 17,562 bp and a large single-copy (LSC) region of 85,745 bp. The plastome contained 129 genes, including 84 protein-coding genes, 37 tRNA genes, and 8 rRNA genes. Most genes had a single copy, while 16 genes were duplicated in the inverted repeat regions.
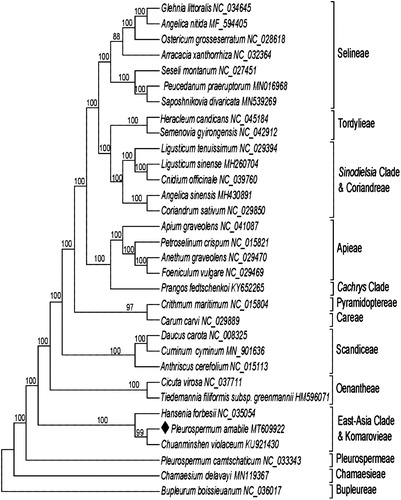
A total of 31 cp genome sequences of Apiaceae were downloaded from the NCBI database to investigate its phylogenetic placement. All sequences were aligned using the MAFFT (Katoh and Standley Citation2013) webserver (http://mafft.cbrc.jp/alignment/server/), a maximum-likelihood (ML) analysis was constructed using RAxML (Stamatakis Citation2014) with 1000 bootstrap replicates, and rooted with Bupleurum boissieuanum (NC_036017). The results showed that P. amabile was closely related to Chuanminshen violaceum (). Meanwhile, the phylogenetic relationships among major clades of Apiaceae were consistent with previous studies (Zhou et al. Citation2020). To better resolve the relationships between Pleurospermum and its allied genera, more data from cp genomes are needed.
Figure 1. Maximum likelihood (ML) tree of 32 species in the family Apiaceae based on the complete chloroplast sequences using Bupleurum boissieuanum (NC_036017) as an outgroup. Numbers on the nodes are bootstrap values from 1000 replicates.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The data that support the findings of this study are openly available in GenBank of NCBI at https://www.ncbi.nlm.nih.gov, reference number MT609922.
Additional information
Funding
References
- Katoh K, Standley D. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30(4):772–780.
- Sheh ML, Pu FD, Pan ZH, Watson MF, Cannon JFM, Holmes-Smith I, Kljuykov EV, Phillippe LR, Pimenov MG, 2005. Apiaceae. In: Flora of China. vol. 14. St. Louis (MO): Missouri Botanical Garden Press; p. 1–205.
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30(9):1312–1313.
- Valiejo-Roman CM, Terentieva EI, Pimenov MG, Kljuykov EV, Samigullin TH, Tilney PM. 2012. Broad polyphyly in Pleurospermum s. l. (Umbelliferae-Apioideae) as inferred from nrDNA ITS and chloroplast sequences. Syst Bot. 37(2):573–581.
- Zhou J, Gao Y-z, Wei J, Liu Z-W, Downie SR. 2020. Molecular phylogenetics of Ligusticum (Apiaceae) based on nrDNA ITS sequences: placement of the Chinese endemic species and a reduced circumscription of the genus. Int J Plant Sci. 181(3):306–323.
- Zhou J, Liu ZW. 2020. The complete chloroplast genome and phylogenetic analysis of Cuminum cyminum. Mitochondrial DNA Part B. 5(1):1079–1080.