
Abstract
Verbascum chinense is a perennial plant in the Scrophulariaceae family that is traditionally used as sedative, astringent, febrifuge, and for skin eruptions. Here, we determined the complete chloroplast(cp) genome sequence for V. chinense using genome skimming sequencing. The cp genome was 153,618 bp and consisted of a large single copy (LSC) region (84,834 bp), a small single copy (SSC) region (17,884 bp), and two inverted repeats (IRs) (25,450 bp). It encodes 114 unique genes, including 80 protein-coding genes, four rRNAs, and 30 tRNAs. Phylogenetic analysis indicates that V. chinense exhibits a closer relationship with V. phoeniceum rather than Scrophularia.
The genus Verbascum (Scrophulariaceae), commonly known as mullein, comprises about 360 species of flowering plants. Verbascum species possess a wide spectrum of biological activities; therefore, they have been used in traditional medicine for centuries (Georgiev et al. Citation2011). Verbascum chinense has conventionally been used as a sedative, an astringent, and a febrifuge as well as to treat skin eruptions (Kaur and Upadhyaya Citation2016). However, the phylogenetic position of V. chinense remains unclear, and there are limited studies on this species, particularly focusing on its genetics. The emergence of next-generation sequencing (NGS) technologies has enabled the rapid use of massive genomic data (He et al. Citation2017; Liu et al. Citation2017). The present study is the first to report the complete plastome of V. chinense. The plastome sequence is deposited in GenBank under the accession number MT610040.
The plant material of V. chinense was collected from Haroshi, Maharashtra, India (17°55′58.68″N, 73°37′36.67″E). The voucher specimen (#LP173132; collected by Pan Li) is stored at the Herbarium of Zhejiang University (HZU). Total genomic DNA was extracted from leaf tissue using Plant DNAzol Reagent (LifeFeng, Shanghai, China) according to the manufacturer’s protocol. High-quality DNA was sheared, and a paired-end library was prepared and sequenced on Illumina HiSeq X10 at Beijing Genomics Institute (BGI, Wuhan, China). The paired-end reads were trimmed and assembled into contigs using the CLC Genomics Workbench 9.5.2 (CLC Inc., Rarhus, Denmark). Next, the complete plastome of V. chinense was constructed using Scrophularia henryi (GenBank accession number: MF861203) as a reference sequence and annotated using Geneious R11 (Biomatters, Auckland, New Zealand), as described by Liu et al. (Citation2018).
The complete plastome of V. chinense is 153,618 bp in length and includes a large single-copy (LSC) region of 84,834 bp, a small single-copy (SSC) region of 17,884 bp, and a pair of inverted repeat (IR) regions (IRa and IRb) of 25,450 bp. The plastome contains 114 unique genes, including 80 protein-coding, 30 tRNA, and four rRNA genes, as well as 20 duplicate genes in the IR regions. Six tRNA and eight protein-coding genes contain a single intron, whereas three genes (rps12, clpP, and ycf3) contain two introns. The overall GC content of the whole plastome is 38.0%, and the GC content of the LSC, SSC, and IR regions is 36.1%, 32.3%, and 43.2%, respectively.
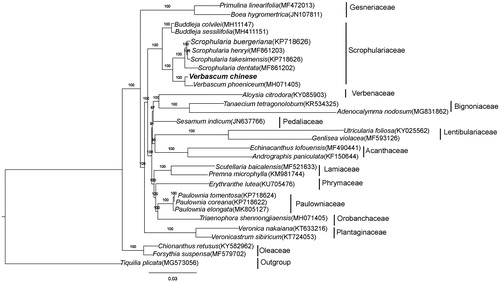
A phylogenetic tree, including V. chinense and 28 other species in Lamiales, was constructed based on whole plastome sequences using the maximum likelihood (ML) method implemented in RAxML-HPC v8.1.11 on the CIPRES cluster (Stamatakis 2014); Tiquilia plicata was used as the outgroup. V. chinense formed a clade with V. phoeniceum and was sister to four Scrophularia species with a 100% bootstrap value ().
Figure 1. Phylogenetic relationships of Lamiales inferred based on whole chloroplast genome sequences. Number above each node indicates the ML bootstrap support values.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The data that support the findings of this study are openly available in GenBank of NCBI at https://www.ncbi.nlm.nih.gov, reference number MT610040.
Additional information
Funding
References
- Georgiev MI, Ali K, Alipieva K, Verpoorte R, Choi YH. 2011. Metabolic differentiations and classification of Verbascum species by NMR-based metabolomics. Phytochemistry. 72(16):2045–2051.
- He Y, Yuan W, Dong M, Han Y, Shang F. 2017. The first genetic map in sweet osmanthus (Osmanthus fragrans lour.) using specific locus amplified fragment sequencing. Front Plant Sci. 8:1621.
- Kaur V, Upadhyaya K. 2016. Antibacterial activity of Verbascum chinense (Scrophulariaceae) extracts. Int J Curr Microbiol App Sci. 5(4):578–584.
- Liu LX, Li P, Zhang HW, Worth J. 2018. Whole chloroplast genome sequences of the Japanese hemlocks, Tsuga diversifolia and T. sieboldii, and development of chloroplast microsatellite markers applicable to East Asian Tsuga. J Forest Res. 23(5):318–323.
- Liu LX, Li R, Worth JRP, Li X, Li P, Cameron KM, Fu CX. 2017. The complete chloroplast genome of Chinese bayberry (Morella rubra, Myricaceae): implications for understanding the evolution of Fagales. Front Plant Sci. 8:968.
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30(9):1312–1313.