
Abstract
Alhagi sparsifolia is a sand-resistant subshrub and food resource for camels in the desert and semi-desert areas of Central Asia. In China, this is the olny Alhagi species and it is restricted in the Northwestern region. Its complete chloroplast genome was sequenced using the Illumina Hiseq X-Ten platform. The genome lacks an inverted repeat (IR) region, containing 74 protein-coding genes, 30 tRNAs genes, and four rRNAs. The overall GC content is 43.6%. Based on the chloroplast genome sequence, a maximum-likelihood (ML) tree was constructed along with its 15 taxa, indicating that A. sparsifolia belong to the tribe Hedysareae, which nested in IRLC group of the subfamily Papilionoideae (Fabaceae).
Alhagi Gagnebin (Fabaceae), contain 3–5 species, naturally distribute in Mediterranean region, Western Asia, Caucasus, Central Asia, Northern South Asia, Himalayas, Mongolia, and Northwest China (Duan Citation2015). In China, Alhagi sparsifolia Schap. is the only species of this genus recorded in the Northwest (Xinjiang, Qinghai, Ningxia, Inner Mongolia, etc.) (Duan et al. Citation2015). As a perennial subshrub with deep strong roots, it can prevent wind and fix sand in desert and oasis (Jin Citation1994; Ma, Li, et al. Citation2018). Its leaves are food sources for camels, its seeds and chemical constituents are often used as medicine in locality (Ma, Shi, et al. Citation2018; Muratova et al. Citation2019). Few study focused on the genome of A. sparsifolia except Wu et al. (Citation2015) made a transcriptomic analysis of its primary roots. A good knowledge in complete chloroplast genomic information of this species would contribute to the study of population genetics, diversity, medical use, and the establishment of efficient protection strategy.
Fresh leaves of the sample were collected from Turpan (Xinjiang, China) and preserved in silica gel. The voucher specimen ‘L.Duan 2016021’ was deposited in the South China Botanical Garden (IBSC). Total genomic DNA was extracted from silica-dried leaves referring to CTAB method (Doyle Citation1987). Then we fragmented them into about 500 bp in size. The genomic library was prepared and sequenced on the Illumina Hiseq X-Ten platform (Illumina Inc., San Diego, CA). The adapters of the raw data were removed by Trimmomatic (Bolger et al. Citation2014). SPAdes version 3.11 (Bankevich et al. Citation2012) was applied for the complete chloroplast (cp) genome de novo assembly. The cp genome was annotated by Dual Organellar GenoMe Annotator (DOGMA) (Wyman et al. Citation2004) and deposited in GenBank (accession number: MT571455).
About 1.33 Gb raw reads were obtained in total, with coverage of 1779× and 128,233 bp in length. The cp genome lacked inverted repeat (IR) region. The genome contained 74 protein-coding genes (CDS), 30 transfer RNA genes (tRNA), four ribosomal RNA genes (rRNA), within which 15 genes (atpF, clpP, ndhA, petB, petD, rpl2, rpl16, rpoC1, rps12, trnA-UGC, trnG-UCC, trnI-GAU, trnK-UUU, trnL-UAA, and trnV-UAC) had one intron, one gene (ycf3) had two introns. Overall GC content of the whole genome was 43.6%.
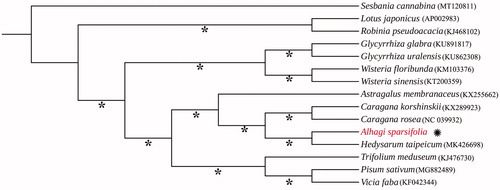
To infer the phylogenetic relationship of this newly sequenced A. sparsifolia with related genera, 14 chloroplast genomes were downloaded from GenBank and were applied to construct the systematic tree. All accession numbers of these download genomes are shown in . Geneious prime and Mauve plugin were used to adjust gene order and remove one copy of IR regions of chloroplast genomes. We aligned these 15 cp genomes using MAFFT version 7 (Katoh and Standley Citation2013) and generated a maximum-likelihood (ML) tree through the program IQ-TREE version1.4.2 (Nguyen et al. Citation2015). The result () showed that Alhagi has a close relationship with Hedysarum, and they undoubtedly belong to the tribe Hedysareae. Broadly speaking, the genus affiliate with the inverted repeat-lacking clade (IRLC) of the subfamily Papilionoideae, which is congruent with the previous study (Amirahmadi et al. Citation2014; Duan et al. Citation2015; Li et al. Citation2020).
Figure 1. Maximum-likelihood phylogenetic tree inferred from 15 chloroplast genomes of Fabaceae. The position of A. sparsifolia is highlighted. The bootstrap values are shown above each node and the values of 100% are shown with asterisks.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The data that support the findings of this study are openly available in GenBank at https://blast.ncbi.nlm.nih.gov/
References
- Amirahmadi A, Osaloo SK, Moein F, Kaveh A, Maassoumi AA. 2014. Molecular systematics of the tribe Hedysareae (Fabaceae) based on nrDNA ITS and plastid trnL-F and matK sequences. Plant Syst Evol. 300(4):729–747.
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, Pyshkin AV, et al. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 19(5):455–477.
- Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 30(15):2114–2120.
- Duan L. 2015. A phylogenetic study on Hedysareae sensu lock (LEGUMINOSAE) [D]. Northwest A&F University. 57.
- Duan L, Wen J, Yang X, Liu PL, Arslan E, Ertuğrul K, Chang ZY. 2015. Phylogeny of Hedysarum and tribe Hedysareae (Leguminosae: Papilionoideae) inferred from sequence data of ITS, matK, trnL-F and psbA-trnH. Taxon. 64 (1):49–64.
- Doyle JJ. 1987. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull. 19:11–15.
- Jin QH. 1994. Alhagi sparsifolia Schap.: a potentially utilizable forage in saline soils.In: Squires VR, Ayoub AT, editors. Halophytes as a resource for livestock and for rehabilitation of degraded lands. Tasks for vegetation science, vol 32, p. 285-288.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30(4):772–780.
- Li DZ, Chen ZD, Wang H, Lu AM, Luo Y, Yu WB. 2020. The families and genera of Chinese vascular plants. Volume II. Beijing: Science Press.
- Ma X, Shi L, Liu X, Zhang D, Wei H. 2018. Preparation of Alhagi sparsifolia Shap monomer compound and screening the antitumor active fractions in vitro. Pharmacol Clin Chinese Materia Medica. 34(4):58–61.
- Ma T, Li X, Lin L, Li L, Li S, Wang B. 2018. The effects of shade on leaf traits and water physiological characteristics in Alhagi sparsifolia. Acta Ecologica Sinica. 38(23):8466–8474.
- Muratova MS, Zou GA, Jenis J, Aisa HA. 2019. Chemical constituents of Alhagi sparsifolia. Chem Nat Compd. 55(5):932–933.
- Nguyen LT, Schmidt HA, von Haeseler A, Minh BQ. 2015. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 32(1):268–274.
- Wu H, Zhang Y, Zhang W, Pei X, Zhang C, Jia S, Li W. 2015. Transcriptomic analysis of the primary roots of Alhagi sparsifolia in response to water stress. PLoS One. 10(3):e0120791.
- Wyman SK, Jansen RK, Boore JL. 2004. Automatic annotation of organellar genomes with DOGMA. Bioinformatics. 20(17):3252–3255.