
Abstract
Sedum emarginatum Migo is a perennial herb endemic to China. Here, we assembled and characterized the complete chloroplast genome of S. emarginatum using Illumina sequencing data. Our assembled chloroplast genome is 149,188 base pairs (bp) in length, containing a large single-copy (LSC) region of 81,399 bp, a small single-copy (SSC) region of 16,721 bp, and two inverted repeat (IR) regions of 25,534 bp. In addition, the chloroplast genome possesses a total of 127 genes, including 82 protein-coding genes, eight rRNA genes, and 37 tRNA genes.
Sedum emarginatum Migo is a perennial herb of the family Crassulaceae (Mort et al. Citation2001). This species is native to China, and can be found in many provinces of southern China. In addition, S. emarginatum is commonly grown as an ornamental and medicinal plant (Bai et al. Citation2019). Here, we assembled and characterized the complete chloroplast genome of S. emarginatum using Illumina sequencing data, providing a valuable genomic resource for future studies.
Fresh leaves of S. emarginatum were collected from Sichuan Province, China (geographic coordinate: 30°37′55.12″ N, 103°10′42.52″ E), and a voucher specimen (ZYC190510) was deposited in the Sichuan University Herbarium (SZ). The total genomic DNA was extracted using a modified CTAB method (Doyle and Doyle Citation1987). One library with an insertion size of ∼300 bp was prepared and sequenced using the Illumina platform. The filtered reads were assembled using NOVOPlasty v2.7.2 (Dierckxsens et al. Citation2017), with the chloroplast genome of Sedum sarmentosum Bunge (Dong et al. Citation2013) as the reference. The assembled chloroplast genome was annotated using Plann v1.1 (Huang and Cronk Citation2015) and manually inspected using Geneious v11.0.3 (Kearse et al. Citation2012). The chloroplast genome together with gene annotations was submitted to GenBank (Sayers et al. Citation2020) under the accession number of MT680404. To further investigate the phylogenetic placement of S. emarginatum, a maximum likelihood tree was constructed by including nine additional Crassulaceae species and one Penthoraceae species. Here, DNA sequences from these 11 chloroplast genomes were first aligned using MAFFT v1.3.13 (Katoh and Standley Citation2013), and a phylogenetic tree was then constructed using RAxML v8.2.11 (Stamatakis Citation2014) with 1000 rapid bootstrap replicates (Stamatakis et al. Citation2008).
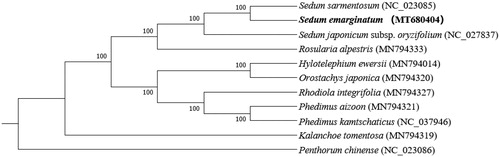
The chloroplast genome of S. emarginatum is 149,188 base pairs (bp) in legnth, containing a large single-copy (LSC) region of 81,399 bp, a small single-copy (SSC) region of 16,721 bp, and two inverted repeat (IR) regions of 25,534 bp. The overall GC-content of the chloroplast genome is 37.8%, and the GC-contents of the LSC, SSC, and IR regions are 35.7%, 31.8%, and 43.0%, respectively. In addition, the chloroplast genome of S. emarginatum possesses a total of 127 genes, including 82 protein-coding genes, eight rRNA genes, and 37 tRNA genes. In addition, our phylogenetic analyses show that S. emarginatum is closely related to S. sarmentosum with a 100% bootstrap support ().
Figure 1. Phylogenetic relationships of Sedum emarginatum based on complete chloroplast genome sequences. Bootstrap percentages and GenBank accession numbers are indicated for each branch and taxon, respectively.
Disclosure statement
No potential conflict of interest was reported by the authors.
Data availability statement
The data that support the finding of this study are open available in NCBI at http://www.ncbi.nlm.nih.gov/, reference number [MT680404], or available from the corresponding author.
Additional information
Funding
References
- Bai LC, Li YL, Duan XM, Yin GJ, Lin Q, Ma QJ, Shi CH, Chen GX. 2019. First report of powdery mildew caused by erysiphe sedi on Sedum emarginatum in China. Plant Dis. 103(9):2474–2474.
- Dierckxsens N, Mardulyn P, Smits G. 2017. NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 45(4):e18.
- Dong WP, Xu C, Cheng T, Lin K, Zhou SL. 2013. Sequencing angiosperm plastid genomes made easy: a complete set of universal primers and a case study on the phylogeny of saxifragales. Genome Biol Evol. 5(5):989–997.
- Doyle JJ, Doyle JL. 1987. A Rapid DNA isolation procedure from small quantities of fresh leaf tissues. Phytochem Bull. 19:11–15.
- Huang DI, Cronk QC. 2015. Plann: a command-line application for annotating plastome sequences. Appl Plant Sci. 3(8):1500026.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30(4):772–780.
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, et al. 2012. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 28(12):1647–1649.
- Mort ME, Soltis DE, Soltis PS, Francisco-Ortega J, Santos-Guerra A. 2001. Phylogenetic relationships and evolution of Crassulaceae inferred from matK sequence data. Am J Bot. 88(1):76–91.
- Sayers EW, Cavanaugh M, Clark K, Ostell J, Pruitt KD, Karsch-Mizrachi I. 2020. GenBank. Nucleic Acids Res. 48(D1):D84–D86.
- Stamatakis A, Hoover P, Rougemont J. 2008. A rapid bootstrap algorithm for the RAxML web servers. Syst Biol. 57(5):758–771.
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30(9):1312–1313.