
Abstract
Phoebe bournei is an endangered Lauraceae species in the world. In this study, the complete chloroplast genome sequence of Phoebe bournei was characterized. Its chloroplast genome size has 152,860 bp, containing a large single copy region of 93,778 bp, a small single copy region of 18,936 bp and a pair of IR regions of 20,073 bp. The overall GC contents of the chloroplast genome sequence was 39.1%. This circular genome contains 127 annotated genes, including 83 PCGs, 36 tRNAs and 8 rRNAs. The phylogenetic tree analysis result showed that Phoebe bournei was the closest relationship with Phoebe chekiangensis using the maximum likelihood (ML) method. This complete chloroplast genomes can be used for the forestry development of this valuable species in the future.
Phoebe bournei is evergreen trees or shrubs belonging to the genus Phoebe the family Lauraceae, which also is an endangered Lauraceae species in the world. The genus Phoebe comprises of approximately 100 species, which distributed in tropical and subtropical Asia and America. The total 34 species and 3 varieties are endemic from China. Phoebe bournei is distributed in Yangtze River Basin and its south region in China (Wu et al. Citation2008). The genus Phoebe species also have a very high economic and ecological value but with extremely limited studies on biochemical compound and population diversity (Zhang et al. Citation2012). However, our understanding of the origins of the chloroplast genome of this forestry plant is limited. In this study, the complete chloroplast genome of Phoebe bournei was characterized and the phylogenetic relationship was explored, which can be used for the forestry development of this valuable species in the future.
The specimen of Phoebe bournei was isolated from Longtan Village Dahe Town test field in Enshi, Hubei, China (109.09E, 29.50 N). The total DNA of Phoebe bournei was extracted using improved CTAB method and stored in Enshi Tujia and Miao Autonomous Prefecture Academy of Forestry (No. ETMAPAF02). The sample total DNA was extracted and the whole genome was sequenced. Quality control was performed to remove low-quality reads and adapters using the FastQC software (Andrews Citation2015). The chloroplast genome was assembled using the SPAdes software (Bankevich et al. Citation2012) and annotated using the Blast and DOGMA software (Wyman et al. Citation2004). The tRNA genes were further identified using the tRNAscan-SE software (Lowe and Chan Citation2016). The annotated chloroplast genome was submitted to GenBank database under accession No.NK823649.
The complete chloroplast genome of Phoebe bournei was a circle with 152,860 bp in size, containing a large single copy region (LSC) of 93,778 bp, a small single copy region (SSC) of 18,936 bp and a pair of inverted repeat regions (IRs) of 20,073 bp. The total 127 genes were annotated on this chloroplast genome, including 83 protein-coding genes (PCG), 36 transfer RNA genes (tRNA) and 8 ribosomal RNA genes (rRNA). On the IR regions, 15 genes were found duplicated, including 5 PCG species (ycf2, ndhB, rps7, rps12 and ycf1), 6 tRNA species (trnL-CAA, trnV-GAC, trnI-GAU, trnA-UGC, trnR-ACG and trnN-GUU) and 4 rRNA species (rrn16, rrn23, rrn4.5 and rrn5). The overall nucleotide composition is: 30.1% of A, 30.8% of T, 19.9 of % C, and 19.2% G, with a total GC content of 39.1%.
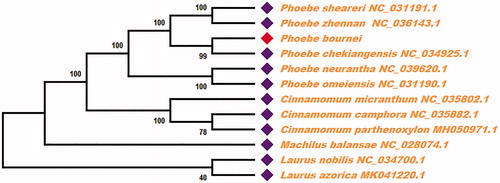
For phylogenetic tree analysis, we selected other 11 plants chloroplast genomes from GenBank to assess the relationship with Phoebe bournei. The genome-wide alignment of all chloroplast genomes was constructed using the MAFFT software (Katoh and Standley Citation2013). The phylogenetic trees were reconstructed using maximum likelihood (ML) methods. ML analysis were performed using the RaxML software (Stamatakis Citation2014), which the bootstrap values were calculated using 2000 replicates to assess node support and all the nodes were inferred with strong support by the ML methods. The final tree was represented and edited using MEGA X (Kumar et al. Citation2018). As shown in of the ML phylogenetic tree, the chloroplast genome of Phoebe bournei was most related to Phoebe chekiangensis, with bootstrap support values of 99%.
Figure 1. The maximum likelihood (ML) tree inferred from Phoebe bournei with other 11 plants chloroplast genomes. The bootstrap support values are shown at the branches. This tree was drawn without setting out groups.
Data openly available in a public repository that issues datasets with DOIs
The data that support the findings of this study are openly available in GenBank of NCBI at https://www.ncbi.nlm.nih.gov, reference number NK823649.
Disclosure statement
The authors have declared that no competing interests exist. This study does not contain any studies with human participants or animals performed by any of the authors.
Data availability statement
The data that support the findings of this study are available from the corresponding author, upon reasonable request.
Additional information
Funding
References
- Andrews S. 2015. FastQC: a quality control tool for high throughput sequence data. http://www.bioinformatics.babraham.ac.uk/projects/fastqc/
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, et al. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 19(5):455–477.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30(4):772–780.
- Kumar S, Stecher G, Li M, Knyaz C, Tamura K. 2018. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 35(6):1547–1549.
- Lowe TM, Chan PP. 2016. tRNAscan-SE On-line: integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 44(W1):W54–W57.
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30(9):1312–1313.
- Wu ZY, Peter HR, Hong DY. 2008. Flora of China. Beijing: Science press.
- Wyman SK, Jansen RK, Boore JL. 2004. Automatic annotation of organellar genomes with DOGMA. Bioinformatics. 20(17):3252–3255.
- Zhang R, Zhou ZC, Jin GQ, Wang SH, Wang XH. 2012. Genetic diversity and diferentiation within three species of the family Lauraceae in southeast China. Biochem Syst Ecol. 44:317–324.