
Abstract
Completed mitochondrial genome of a new species candidate of Rosa rugosa, named as Rosa angusta, is 303,484 bp long. The overall GC content of this mitochondrial genome is 45.2%. It contains 52 genes covering 31 protein-coding genes, 17 tRNAs, and 3 rRNAs. In comparison to R. rugosa mitochondrial genome assembled from the public NGS raw reads, 124 SNPs and 769 INDELs were identified. Phylogenetic trees suggest that more Rosa mitochondrial genomes will be needed to understand phylogenetic relationship of the two Rosa species.
Rosa rugosa, one of the firstly reported Rosa species in Korea, is distributed in Korea, Japan, China, and Russia (Rehder Citation1949). One population of R. rugosa was identified in 2013 by Suhwan Nam, one of the authors, showing significant differences of leaves and flowers (Kim, Heo, et al. Citation2019), named as Rosa augusta (Kim, Park, et al. Citation2019). Its chloroplast genome was successfully deciphered presenting 40 single nucleotide polymorphisms (SNPs) and 224 insertions and deletions (INDELs) between R. augusta and Chinese R. rugosa (Kim, Heo, et al. Citation2019). For understanding genetic background of this species, we successfully assembled its complete mitochondrial genome.
Its total DNA isolated from Hagampo coast, Wonbuk-myeon, Taean-gun, Chungcheongnam-do, Republic of Korea, was extracted from fresh leaves by using a DNeasy Plant Mini Kit (QIAGEN, Hilden, Germany). Voucher was deposited in InfoBoss Cyber Herbarium (IN; IB-90006). Genome was sequenced using HiSeqX at Macrogen Inc., Korea, and de novo assembly and gap-filling process were performed by Velvet v1.2.10 (Zerbino and Birney Citation2008) and SOAPGapCloser v1.12 (Zhao et al. Citation2011), respectively, and confirmation of each assembled bases and correctly assembled sequences done by BWA v0.7.17 (Li Citation2013), and SAMtools v1.9 (Li et al. Citation2009) under the environment of Genome Information System (GeIS; http://geis.infoboss.co.kr/; Park et al. in preparation). In addition, circular test was conducted for confirming that our mitochondrial genome is circular form using SOAPGapCloser v1.12 in the same way used in completing bacterial genome (Park et al. Citation2020). Mitochondrial genome annotation was conducted with Mitofy (Alverson et al. Citation2010) and then annotated genes were confirmed by comparing with mitochondrial genome of Rosa chinensis (PDCK01000047) under the environment of Geneious R11 11.0.5 (Biomatters Ltd., Auckland, New Zealand).
The mitochondrial genome of R. angusta (GenBank accession is MN909970) is 303,484 bp (GC ratio is 45.2%). It contains 52 genes (31 protein-coding genes, 3 rRNAs, and 17 tRNAs). Simple sequence repeats (SSRs) were identified using the pipeline of the SSR database (SSRDB; http://ssr.pe.kr/; Park et al. in preparation) which has been utilized in various studies (Kim, Park, et al. Citation2019; doi:10.1093/jisesa/ieaa090; Lee et al. accepted; https://www.hindawi.com/journals/ijg/2020/3236461). In total, 1,051 SSRs of which total length is 11,706 bp (3.86%) were identified. 709 pentaSSRs and 199 hexaSSRs (86.39% in total) are classified as potential SSRs, which is similar to those of three Dysphania species (around 80%; Kim, Park, et al. Citation2019).
We also assembled Rosa rugosa mitochondrial genome from the public NGS raw reads (SRA accession is SRR7077019; Saint-Oyant et al. Citation2018), displaying that its length is 302,831 bp and 52 genes with the same configuration of those of R. angusta (GenBank accession is BK013300). Numbers of single nucleotide polymorphisms (SNPs) and insertions and deletions (INDELs) between two mitochondrial genomes are 124 (0.041%) and 769 (0.25%), respectively. These numbers are larger than those between two Malus × domestica mitochondrial genomes (NC_018554 and MN964891; Goremykin et al. Citation2012; Ge et al. Citation2020), suggesting that genetic distance between two Rosa mitochondrial genomes can be considered as interspecific relation. It is also consistent in those of Bryophyte species, including Marchandia polymorpha subsp. ruderalis (Kwon et al. Citation2019), Riccia fluitans (Min et al. Citation2020), and Monosolenium tenerum (Dong et al. Citation2019). However, additional investigations are required because some of intraspecific variations on mitochondrial genomes, including Liriodendron tulifipera (Park et al. Citation2019) and Arabidopsis thaliana (Park et al. in preparation) display larger numbers.
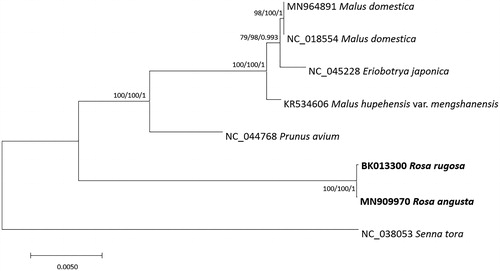
Seven complete mitochondrial genomes in Rosaceae including those of R. angusta and R. rugosa assembled in this study and that of Senna tora (Fabaceae) as outgroup species, were used for constructing neighbor-joining (bootstrap repeat is 10,000), maximum likelihood (bootstrap repeat is 1,000), and Bayesian Inference (Number of generation is 1,100,000) phylogenic trees using MEGA X (Kumar et al. Citation2018) and MrBayes 3.2.7a (Ronquist et al. 2012) after aligning 21 conserved genes using MAFFT v7.450 (Katoh and Standley Citation2013) and concatenating these alignments. We excluded one mitogenome of R. chinensis because its gene annotation is different from those of the other mitogenomes in Rosaceae. Phylogenetic trees show that two Rosa species are clustered together (). In addition, position of Eriobotrya japonica is supported by slightly lower supportive values of three phylogenetic trees (), which is congruent to the phylogenetic analysis using its whole genome (Jiang et al. Citation2020). Additional mitochondrial genomes of Rosa species will provide better result to understand phylogenetic relationship of these two Rosa species using mitochondrial genomes. This complete mitochondrial genome supports R. angusta is a new species together with its chloroplast genome sequences (Kim, Heo, et al. Citation2019).
Figure 1. Neighbor-joining (bootstrap repeat is 10,000), maximum likelihood (bootstrap repeat is 1,000), and Bayesian Inference (Number of generations is 1,100,000) phylogenetic trees of 21 conserved genes (atp1, atp4, atp6, atp9, ccmB, ccmC, ccmFN, cob, cox1, cox2, matR, nad1, nad2, nad3, nad4, nad5, nad6, nad7, nad9, rps1, and rps12) originated from the seven mitochondrial genomes of Asteraceae and one that of Fabaceae: Rosa angusta (MN909970 in this study), Rosa rugosa (BK013300), Malus domestica (NC_018554 and MN964891; CitationGoremykin et al. 2012; CitationGe et al. 2020), Malus hupehensis var. mengshanensis (KR534606; Duan et al. Citation2016), Eriobotrya japonica (NC_045228), Prunus avium (NC_044768; CitationYan et al. 2019), and Senna tora (NC_038053) as outgroup species. Phylogenetic tree was drawn based on maximum likelihood tree. The numbers above branches indicate bootstrap support values of maximum likelihood, neighbor-joining, and Bayesian Inference phylogenetic trees, respectively.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The mitochondrial genome in this study can be accessed via NCBI GenBank (https://www.ncbi.nlm.nih.gov/genbank/) with the accession numbers of MN909970.
Additional information
Funding
References
- Alverson AJ, Wei X, Rice DW, Stern DB, Barry K, Palmer JD. 2010. Insights into the evolution of mitochondrial genome size from complete sequences of Citrullus lanatus and Cucurbita pepo (Cucurbitaceae). Mol Biol Evol. 27(6):1436–1448.
- Dong S, Zhao C, Zhang S, Zhang L, Wu H, Liu H, Zhu R, Jia Y, Goffinet B, Liu Y. 2019. Mitochondrial genomes of the early land plant lineage liverworts (Marchantiophyta): conserved genome structure, and ongoing low frequency recombination. BMC Genomics. 20(1):953
- Duan N, Sun H, Wang N, Fei Z, Chen X. 2016. The complete mitochondrial genome sequence of Malus hupehensis var. pinyiensis. Mitochondrial DNA Part A. 27(4):2905–2906.
- Ge D, Dong J, Guo L, Yan M, Zhao X, Yuan Z. 2020. The complete mitochondrial genome sequence of cultivated apple (Malus domestica cv.‘Yantai Fuji 8’). Mitochondrial DNA Part B. 5(2):1317–1318.
- Goremykin VV, Lockhart PJ, Viola R, Velasco R. 2012. The mitochondrial genome of Malus domestica and the import-driven hypothesis of mitochondrial genome expansion in seed plants. Plant J. 71(4):615–626.
- Jiang S, An H, Xu F, Zhang X. 2020. Chromosome-level genome assembly and annotation of the loquat (Eriobotrya japonica) genome. GigaScience. 9(3):giaa015.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30(4):772–780.
- Kim Y, Heo K-I, Nam S, Xi H, Lee S, Park J. 2019. The complete chloroplast genome of candidate new species from Rosa rugosa in Korea (Rosaceae.). Mitochondrial DNA Part B. 4(2):2433–2435.
- Kim Y, Park J, Chung Y. 2019. Comparative analysis of chloroplast genome of Dysphania ambrosioides (L.) Mosyakin & Clemants understanding phylogenetic relationship in genus Dysphania R. Br. Korean J Plant Resource. 32(6):644–668.
- Kumar S, Stecher G, Li M, Knyaz C, Tamura K. 2018. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 35(6):1547–1549.
- Kwon W, Kim Y, Park J. 2019. The complete mitochondrial genome of Korean Marchantia polymorpha subsp. ruderalis Bischl. & Boisselier: inverted repeats on mitochondrial genome between Korean and Japanese isolates. Mitochondrial DNA Part B. 4(1):769–770.
- Lee J+, Park J+, Kwon W, Xi H, Park J*, The complete mitochondrial genome of Figulus binodulus Waterhouse, 1873 (Coleoptera: Lucanidae), Journal of Insect Science, accepted.
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 2009. The sequence alignment/map format and SAMtools. Bioinformatics. 25(16):2078–2079.
- Li H. 2013. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv (preprint arXiv:13033997).
- Min J, Kwon W, Xi H, Park J. 2020. The complete mitochondrial genome of Riccia fluitans L.(Ricciaceae, Marchantiophyta): investigation of intraspecific variations on mitochondrial genomes of R. fluitans. Mitochondrial DNA Part B. 5(2):1220–1222.
- Park J, Kim Y, Kwon M. 2019. The complete mitochondrial genome of tulip tree, Liriodendron tulipifera L. (Magnoliaceae): intra-species variations on mitochondrial genome. Mitochondrial DNA Part B. 4(1):1308–1309.
- Park J, Xi H, Park J, Name S-J, Lee Y-D. 2020. Complete Genome Sequence of the Blochmannia Endosymbiont of Camponotus nipponensis. Microbiol Resource Announcement. 9(29):e00703–20.
- Rehder A. 1949. Bibliography of cultivated trees and shrubs hardy in the cooler temperate regions of the northern hemisphere. https://www.biodiversitylibrary.org/item/122372#page/4/mode/1up
- Ronquist, F., Teslenko, M., Van Der Mark, P., Ayres, D.L., Darling, A., Höhna, S., Larget, B., Liu, L., Suchard, M.A. and Huelsenbeck, J.P., 2012. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Systematic biology, 61(3), 539–542.
- Saint-Oyant HL, Ruttink T, Hamama L, Kirov I, Lakhwani D, Zhou NN, Bourke PM, Daccord N, Leus L, Schulz D, et al. 2018. A high-quality genome sequence of Rosa chinensis to elucidate ornamental traits. Nat Plants. 4(7):473–484.
- Yan M, Zhang X, Zhao X, Yuan Z. 2019. The complete mitochondrial genome sequence of sweet cherry (Prunus avium cv.‘summit’). Mitochondrial DNA Part B. 4(1):1996–1997.
- Zerbino DR, Birney E. 2008. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18(5):821–829.
- Zhao Q-Y, Wang Y, Kong Y-M, Luo D, Li X, Hao P. 2011. Optimizing de novo transcriptome assembly from short-read RNA-Seq data: a comparative study. BMC Bioinf. 12(14):S2.