
Abstract
Salix cardiophylla was a member of the genus of Salix in family Salicaceae with unique morphological traits, and once recognized as a separate genus, Toisusu Kimura. Here, we sequenced and assembled the complete mitochondrial genome of S. cardiophylla, which was 735,173 bp in length, including 56 genes, 28 protein-coding genes, 3 rRNA genes, 25 tRNA genes, and one large inverted repeat regions with length of 13,603 bp. Phylogenetic analysis based on 26 mitochondrial CDS confirmed that S. cardiophylla is a member of Salix, and support its merge into Salix in aspect of our new insights on mitogenome phylogenomics.
Salix cardiophylla Trautv. & C.A.Mey. is deciduous tree up to 20 m high found in north to central Honshu and southern Kurils of Japan, growing along the gravel river. Salix cardiophylla belongs to the genus Salix (family Salicaceae), because of its unique reproductive organs, such as the pendulous female catkins and the deciduous styles, it was once treated as a separate genus, i.e., Toisusu Kimura (Barkalov and Kozyrenko Citation2014). The genus Toisusu was then merged into Salix afterwards based on plastid and nuclear sequence evidence (Azuma et al. Citation2000; Ohashi Citation2001; Chen et al. Citation2010). However, merge of Toisusu into Salix based on phylogentic inference of mitochondrial sequence is lack.
Here, we used a modified CTAB method (Sahu et al. Citation2012), the complete mitochondrial genomic DNA was extracted from young leaves of S. cardiophylla which grown in Honshu, Japan (36°N, 138°E). Voucher specimens were stored in the Herbarium of Kunming Institute of Botany (Accession no: T. Tanaka s.n., cult.). The genomic library for Illumina paired-end (PE) sequencing was constructed using the Illumina Hiseq X Ten sequencer. A total of ca. 31.2 million generated and this sequence has been deposited in the NCBI Sequence Read Archive (SRA) with accession number SRR12534676. MiSeq raw reads were assembled via NOVOPlasty v2.7.2 with a k-mer of 39 (Dierckxsens et al. Citation2016). Taking the S. paraflabellaris mitogenome as the reference sequence (Chen et al. Citation2019; Huang et al. Citation2019), the annotation of genome assembled was carried out with Geneious v2020.1.1 software (Kearse et al. Citation2012).
The high-quality annotated mitogenome sequences of S. cardiophylla were deposited in the GenBank database under the accession No. MT806745. The mitochondrial genome of S. cardiophylla was assembled as a closed-circular molecule of 7,35,173 bp in length. It contains 28 protein-coding genes, including ones for NADH dehydrogenase (nad3, 4, 4 L, 6, 7, 9), succinate dehydrogenase (sdh4), apocytochrome b (cob), cytochrome c oxidase (cox1, 2, 3), ATP synthase (atp1, 4, 6, 8, 9), cytochrome c biogenesis (ccmB, C, Fn, Fc), ribosomal proteins (rpl2, 16 and rps1, 3, 4, 7, 12), maturase and membrane transporter (matR). Meantime, the genome contains 25 tRNA genes, and 3 rRNA genes are annotated. The overall base composition is 27.5% A, 22.5% C, 22.3% G, and 27.7% T, and the AT content is higher than GC content. Furthermore, we used the Repeat Finder implemented in Geneious and identified two large repeats with length of 13,603 bp. Similar to the mitogenome of S. paraflabellaris, no genes or protein-coding sequences harbored in the two repeats.
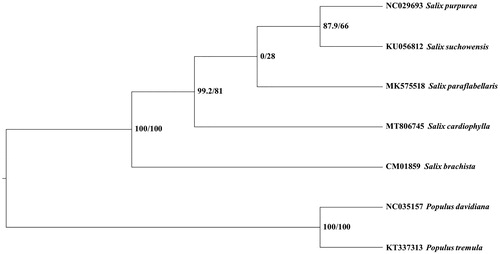
To explore the controversial systematic position of S. cardiophylla, four Salix species and two outgroup Populus species with mitogenome sequence were downloaded from GenBank (Kersten et al. Citation2016; Wei et al. Citation2016; Ye et al. Citation2017; Choi et al. Citation2017; Chen et al. Citation2019; Huang et al. Citation2019). Ultimately, 26 mitochondrial CDS shared by all these seven species were aligned by software MAFFT v7.47 (Katoh and Standley Citation2013). After manual adjustment, the maximum likelihood (ML) tree was constructed using the software IQ_TREE 1.6.2 (Nguyen et al. Citation2015) based on an array with the aligned seven sequences, branch support was estimated by bootstrap value and SH-like approximate likelihood ratio (SHAlrt) (Guindon et al. Citation2010) with 10,000 replicates under HKY + F model according to Bayesian information criterion by the software Model Finder (Kalyaanamoorthy et al. Citation2017). Our ML revealed that five Salix species formed a robust monophyletic clade, confirmed that S. cardiophylla is a member of Salix and support the treatment of merging it back into Salix based on our new insights from mitogenome phylogenomic inference (). This work provides a valuable source of data for the study of the controversial systematic position of S. cardiophylla.
Figure 1. ML phylogenetic tree of S. cardiophylla and six Salicaceae species based on CDS shared by all these seven mitochondrial complete genome, branch supports values were reported as SH-aLRT/UFBoot.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The data that support the findings of this study are openly available in NCBI at https://www.ncbi.nlm.nih.gov, reference number MT806745, SRR12534676.
Additional information
Funding
References
- Azuma T, Kajita T, Yokoyama J, Ohashi H. 2000. Phylogenetic relationships of Salix (Salicaceae) based on rbcl sequence data. Am J Bot. 87(1):67–75.
- Barkalov V, Kozyrenko M. 2014. Phylogenetic analysis of the far eastern Salix (Salicaceae) based on sequence data from chloroplast DNA regions and its of nuclear ribosomal DNA. Bot Pac. 3(1):3–19.
- Chen JH, Huang Y, Brachi B, Yun QZ, Zhang W, Lu W, Li HN, Li WQ, Sun XD, Wang GY, et al. 2019. Genome-wide analysis of cushion willow provides insights into alpine plant divergence in a biodiversity hotspot. Nat Commun. 10(1):5230.
- Chen JH, Sun H, Wen J, Yang YP. 2010. Molecular phylogeny of Salix l. (Salicaceae) inferred from three chloroplast datasets and its systematic implications. Taxon. 59(1):29–37.
- Choi MN, Han M, Lee H, Park H, Kim M, Kim J, Na Y, Sim S, Park E. 2017. The complete mitochondrial genome sequence of Populus davidiana dode. Mitochondrial DNA Part B. 2(1):113–114.
- Dierckxsens N, Mardulyn P, Smits G. 2016. Novoplasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 45:e18.
- Guindon S, Dufayard J, Lefort V, Anisimova M, Hordijk W, Gascuel O. 2010. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of phyml 3.0. Syst Biol. 59(3):307–321.
- Huang Y, Chen X, Chen KY, Li WQ, Shi MM, Chen JH. 2019. The complete mitochondrial genome of Salix paraflabellaris, an endemic alpine plant of Yunnan province of China. Mitochondrial DNA Part B. 4(1):1394–1395.
- Kalyaanamoorthy S, Minh BQ, Wong TKF, Von Haeseler A, Jermiin LS. 2017. Modelfinder: fast model selection for accurate phylogenetic estimates. Nat Methods. 14(6):587–589.
- Katoh K, Standley DM. 2013. Mafft multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30(4):772–780.
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, et al. 2012. Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 28(12):1647–1649.
- Kersten B, Rampant PF, Mader M, Paslier ML, Bounon R, Berard A, Vettori C, Schroeder H, Leple J, Fladung M. 2016. Genome sequences of Populus tremula chloroplast and mitochondrion: implications for holistic poplar breeding. PLoS One. 11(1):e0147209.
- Nguyen L, Schmidt HA, Von Haeseler A, Minh BQ. 2015. Iq-tree: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 32(1):268–274.
- Ohashi H. 2001. Salicaceae of Japan. Sci Rep Tohoku Univ Ser 4 Biol. 40:269–396.
- Sahu SK, Thangaraj M, Kathiresan K. 2012. DNA extraction protocol for plants with high levels of secondary metabolites and polysaccharides without using liquid nitrogen and phenol. ISRN Mol Biol. 2012:205049–205049.
- Wei S, Wang X, Bi C, Xu Y, Wu D, Ye N. 2016. Assembly and analysis of the complete Salix purpurea l. (Salicaceae) mitochondrial genome sequence. Springerplus. 5(1):1894–1894.
- Ye N, Wang X, Li J, Bi C, Xu Y, Wu D, Ye Q. 2017. Assembly and comparative analysis of complete mitochondrial genome sequence of an economic plant Salix suchowensis. PeerJ. 5:e3148