
Abstract
The BS-C-1 cell line widely used in virological research was reportedly derived from the African green monkey Cercopithecus (Chlorocebus) aethiops. We used Sanger sequencing to determine the full nucleotide sequence of mtDNA in BS-C-1 cells. The mitochondrial genome in this cell line is 16,456 bp long and has an organization typical of that in other mammalian genomes with 37 genes, including 2 rRNAs, 22 tRNAs, and 13 polypeptide-encoding genes. Surprisingly, this genome is most closely related to that of Chlorocebus pygerythrus, which does not agree with the reported derivation of this cell line from C. aethiops. Another unexpected observation is the presence of mtDNA length heteroplasmy within the MT-ND6 gene, with the variant containing a stretch of 8 Cs encoding a functional gene, whereas the variant containing 9 Cs encodes a frameshifted gene. It is unclear whether the latter variant is nonfunctional or whether it is corrected by programmed translational ribosomal −1 frameshifting. The availability of the full mtDNA sequence for the BS-C-1 cell line should increase its utility by enabling studies on mtDNA transcription and replication.
BS-C-1 is a popular cell line in virological research (Piccinoti et al. 2913; Frey et al. Citation2017; Ruedas et al. Citation2017) that was derived around 1961 from commercially supplied healthy kidney tube cultures of Cercopithecus aethiops (today Chlorocebus aethiops) (Hopps et al. Citation1963). To expand the utility of this cell line, we determined the full nucleotide sequence of its mitochondrial DNA (mtDNA).
BS-C-1 cells were obtained from the Cell Culture Facility, Biosciences Divisional Services, University of California, Berkeley. A near-complete mitochondrial genome was amplified using GoTaq® Long PCR Master Mix (Promega, Madison, WI) and primers BSC1F1 (CCGAGGACTTCAAACACTACTT) and BSC1R1 (CTGCTTGGTTGCCTCATTTG) designed against Chlorocebus aethiops mtDNA. The PCR product was sequenced directly after cleanup with ExoSap-it reagent (Thermo Fisher Scientific, Waltham, MA) using a set of sequencing primers specific for C. aethiops mtDNA. Analysis of the resulting sequences revealed that mtDNA in BS-C-1 cells most closely resembles that of Chlorocebus pygerythrus. Therefore, a new set of sequencing primers was designed against C. pygerythrus mtDNA sequence and used to complete the sequence of the large fragment. Regions refractory to direct sequencing of PCR products were cloned into pBluescriptII SK + vector, and sequenced as part of the plasmid, with plasmid-specific primers. Finally, a gap in the sequence was closed by amplifying an 860 bp fragment with primers BSCgapF (CCTTCAAATCACCTTAATTT) and BSCgapR (AAGAGAATGAGGCTGTAATC) and sequencing as described above. The final sequence was annotated with MITOS2 (Bernt et al. Citation2013) and deposited with GenBank (accession number MT481926).
Remarkably, one of the areas refractory to direct sequencing of PCR products is located in the MT-ND6 gene. Sequencing of plasmids containing cloned PCR products encompassing this area revealed 8/9 nucleotides length heterogeneity of the polyC stretch between nucleotides 13945–13953. The majority of the cloned products contained a stretch of 9 Cs (confirmed by sequencing of both strands), whereas a minor fraction of cloned fragments contained 8 Cs. This heterogeneity provides a plausible explanation for the difficulties encountered with the direct sequencing of PCR products. This observation also suggests a length heteroplasmy of mtDNA in BS-C-1 cells. Functionally, a 9-nucleotide stretch of Cs results in a frameshift and is predicted to prevent proper translation of the full-length MT-ND6. In contrast, transcripts from mitochondrial genomes containing an 8-nucleotide stretch inside MT-ND6 are predicted to translate without complications. It is unclear whether mitochondrial function in BS-C-1 cells is supported exclusively by genomes containing a stretch of 8 Cs, or whether they are functionally augmented by a programmed ribosomal −1 frameshift during translation of MT-ND6 transcripts containing stretches of 9 Cs.
It is conceivable that co-amplification of the nuclear mitochondrial sequences (NUMTs) containing an alternative number of repeats can manifest itself as mtDNA length heteroplasmy. However, three considerations make this possibility unlikely:
MtDNA, unlike specific NUMTs is present is tens of copies per cell. Assuming that NUMTs and mtDNA are amplified with the same efficiency, the abundance of NUMTs in the final PCR product is expected to be much smaller than that of mtDNA amplification product. As we indicated earlier, we were unable to sequence the nearly-genomic length PCR product (15,843 bp) through this repeat region because of mixed sequence. However, it is known that Sanger sequencing (implemented without special accommodations as in our case) can only detect a variant sequence if its abundance is >20% (Davidson et al. Citation2012). Therefore, we should not have noticed interference from NUMTs, and sequencing difficulties were likely due to a bona fide mtDNA sequence length heteroplasmy.
In order to amplify with primers used in this study, NUMT has to be near-genomic length. All near-genomic-length NUMTs that we are aware of differ in sequence from the resident mtDNA. Therefore, if, for some reason, NUMTs were preferentially amplified so that their abundance increased to >20% and became detectable by Sanger sequencing, we should have also detected heteroplasmy at other loci, which was not the case.
Finally, for a 15,843 bp NUMT to be amplifiable, at least three conditions should be met: a) the NUMT should have a near-genomic length (these are outnumbered by shorter NUMTs in all known genomes. Shorter NUMTs cannot be amplified with long-range primers used in this study), b) our primers should have sufficient homology to NUMTs, and c) the breaking point for NUMT insertion should have occurred within the short 613 bp region between our primers for long-range amplification, which is unlikely. Collectively, these considerations led us to believe that mtDNA length heteroplasmy in BS-C-1 cell line does not result from NUMT contribution.
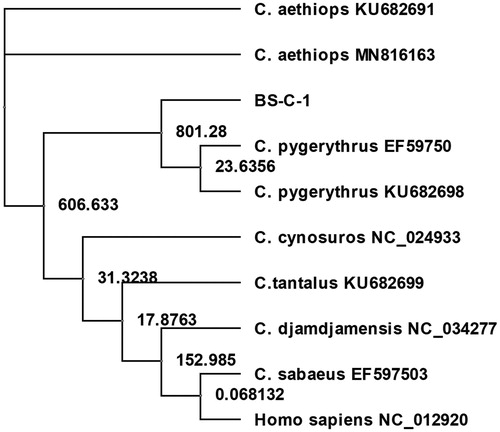
To examine the phylogenetic relationships of the mitochondrial genome in BS-C-1 cells, it was aligned with available mtDNA sequences from representatives of the genus Chlorocebus (C. aethiops (GenBank KU682691 and MN816163), C. tantalus (GenBank KU682699), C. pygerythrus (GenBank KU682698 and EF597501), C. sabaeus (GenBank EF597503), C. cynosuros (GenBank NC_024933) and C. djamdjamensis (GenBank NC_034277)), as well as with the revised Cambridge Reference Sequence of human mtDNA (GenBank NC_012920) using the MUSCLE algorithm of the Unipro UGENE package (Okonechnikov et al. Citation2012). The phylogenetic tree was built using PhyML Maximum Likelihood method (Guindon et al. Citation2010) within the same package (). This analysis confirmed that BS-C-1 mtDNA most closely resembles that of C. pygerithrus, which does not agree well with the reported derivation of this cell line from C. aethiops.
Figure 1. Maximum-likelihood phylogenetic tree showing the relationships of the BS-C-1 mitochondrial genome to other genomes in the genus Chlorocebus and to human mtDNA.
Acknowledgements
The authors would like to express their gratitude to Viktoriya Pastukh and Sunil Mitta for their technical assistance.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The complete nucleotide sequence of the BS-C-1 mtDNA has been deposited in the NCBI GenBank database at (https://www.ncbi.nlm.nih.gov) under the accession number MT481926.
Additional information
Funding
References
- Bernt M, Donath A, Juhling F, Externbrink F, Florentz C, Fritzsch G, Putz J, Middendorf M, Stadler PF. 2013. MITOS: improved de novo metazoan mitochondrial genome annotation. Mol Phylogenet Evol. 69(2):313–319.
- Davidson CJ, Zeringer E, Champion KJ, Gauthier MP, Wang F, Boonyaratanakornkit J, Jones JR, Schreiber E. 2012. Improving the limit of detection for Sanger sequencing: a comparison of methodologies for KRAS variant detection. Biotechniques. 53(3):182–188.
- Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. 2010. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 59(3):307–321.
- Frey TR, Lehmann MH, Ryan CM, Pizzorno MC, Sutter G, Hersperger AR. 2017. Ectromelia virus accumulates less double-stranded RNA compared to vaccinia virus in BS-C-1 cells. Virology. 509:98–111.
- Hopps HE, Bernheim BC, Nisalak A, Tjio JH, Smadel JE. 1963. Biologic characteristics of a continuous kidney cell line derived from the African green monkey. J Immunol. 91:416–424.
- Piccinotti S, Kirchhausen T, Whelan SP. 2013. Uptake of rabies virus into epithelial cells by clathrin-mediated endocytosis depends upon actin. J Virol. 87(21):11637–11647.
- Okonechnikov K, Golosova O, Fursov M, Team U. 2012. Unipro UGENE: a unified bioinformatics toolkit. Bioinformatics. 28(8):1166–1167.
- Ruedas JB, Ladner JT, Ettinger CR, Gummuluru S, Palacios G, Connor JH. 2017. Spontaneous mutation at amino acid 544 of the Ebola virus glycoprotein potentiates virus entry and selection in tissue culture. J Virol. 91(15):e00392–17