
Abstract
Fraxinus malacophylla is one of the commonly used ecological restoration tree species in rocky desertification areas. It has high medicinal and timber value. And has high marketization prospects. The complete chloroplast genome sequence of F. malacophylla was generated by de novo assembly using whole-genome next generation sequencing. The complete chloroplast genome of F. malacophylla was 155621 bp in total sequence length and divided into four distinct regions: large single copy region (86404 bp), small single copy region (17821 bp), and a pair of inverted repeat regions (25698 bp). The F. malacophylla chloroplast genome annotation predicted a total of 131 genes, consisting of 35 tRNA genes, 8 rRNA genes, and 88 protein-coding genes. Phylogenetic analysis with the reported chloroplast genomes revealed that F. malacophylla has most closely related to F. excelsior.
Fraxinus malacophylla is a deciduous tree, which mainly distributed in mountains secondary forest dominated by limestone in Yunnan and Guangxi, China (Huang et al. Citation2019). Its a commonly associated tree species for ecological restoration in rocky desertification areas (Huang and Chen Citation2014). Fraxinus malacophylla has high medicinal and timber value and usually be used to treat constipation, malaria, epilepsy and make furniture (Tan et al. Citation2013; Huang and Chen Citation2014). However, there are no researches about the Fraxinus chloroplast. Therefore, we first assembled the complete chloroplast genome of F. malacophylla, which provides a genomic resource and to clarify the phylogenetic relationship of this plant with other species in the Oleaceae family.
Leaf samples of F. malacophylla were collected from Kunming, Yunnan, China (geospatial coordinates: E102°46′40″, N25°03′07″, altitude: 1954 m). The voucher specimen is deposited at the Southwest Forestry University Herbarium (specimen number: SWFC0048721). The total DNA was isolated using the Plant Genomic DNA Kit. Then, the Illumina Hiseq X Ten platform was used to sequence the DNA (Sangon Biotech (Shanghai) Co. Shanghai, China). The complete cp genomes were assembled by SPAdes (Bankevich et al. Citation2012), Using GapFiller to add GAP to the contig obtained by stitching (Boetzer and Pirovano Citation2012), which compared with the chloroplast sequence of F. xanthoxyloides as a reference. PrInSeS-G was used for sequence correction to correct editing errors and missing insertion of small fragments in the process of splicing (Massouras et al. Citation2010). The genes in the chloroplast genome were predicted using Prokka (Seemann Citation2014) and corrected by Blast search. Simple sequence repeat (SSR) motifs were investigated using RepeatMasker (Altschul et al, Citation1997). The CpDNA sequence of F. malacophylla was submitted to GenBank (accession number: MT663306).
The chloroplast genome of F. malacophylla was a circular form of 1,55,621 bp in length, which was composed of four distinct regions such as large single copy (LSC) region of 86,404 bp, small single copy (SSC) region 17,821 bp and a pair of IRs regions of 25698. The overall GC content was 37.9% and the GC contents of the LSC, SSC, and IR regions are 35.9%, 32.1%, and 43.2%, respectively. The chloroplast genome contained a total of 133 genes, including 88 protein coding genes, 35 tRNA genes and 8 rRNA genes.
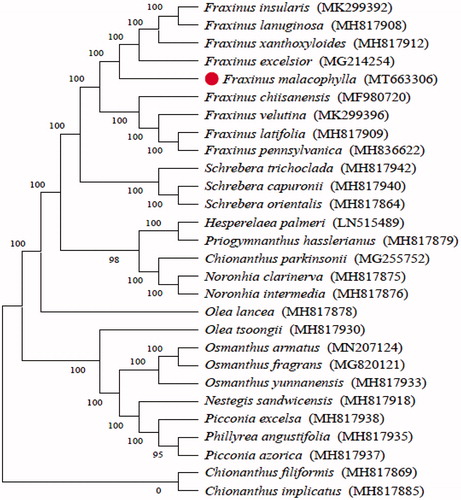
In order to understand the phylogenetic relationship between F. malacophylla and related species, the complete chloroplast genome sequences of 13 genera (28 species) were aligned by MAFFT (Katoh et al. Citation2002) and trimmed properly by trimAl (Capella-Gutierrez et al. Citation2009). The evolutionary history was inferred by using the Maximum Likelihood method based on the Tamura-Nei model in MEGA7.0 (Kumar et al. Citation2016). Bootstrap (BS) values were calculated from 1000 replicate analyses (). As was expected, F. malacophylla was placed within Apiaceae tribe Fraxineae, and comprise a clade with F. insularis, F. lanuginosa, F. xanthoxyloides and F. excelsior subsp. greenmannii with 100% BS value.
Figure 1. The maximum likelihood phylogenetic tree constructed from 28 species chloroplast genomes. The numbers at each node indicate bootstrap support.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability
The data that support the findings of this study are openly available in [National Center for Biotechnology Information], [https://www.ncbi.nlm.nih.gov/], accession number [MT663306].
Additional information
Funding
References
- Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25(17):3389–3402.
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, et al. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 19(5):455–477.
- Boetzer M, Pirovano W. 2012. Toward almost closed genomes with GapFiller. Genome Biol. 13(6):R56.
- Capella-Gutierrez S, Silla-Martinez JM, Gabaldon T. 2009. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics. 25(15):1972–1973.
- Huang CL, Chen Q. 2014. Afforestation technology of Fraxinus malacophylla in semi-arid subtropical rocky desertification area. For Sci Technol. 39(12):20–22.
- Huang JW, Sun YL, Zhou JX, Liu YG, Wan L. 2019. Growth and photosynthetic characteristic responses of Fraxinus malacophylla to different soil moisture conditions. J Zhejiang A F Univ. 36(06):1254–1260.
- Katoh K, Misawa K, Kuma K, Miyata T. 2002. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30(14):3059–3066.
- Kumar S, Stecher G, Tamura K. 2016. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 33(7):1870–1874.
- Massouras A, Hens K, Gubelmann C, Uplekar S, Decouttere F, Rougemont J, Cole ST, Deplancke B. 2010. Primer-initiated sequence synthesis to detect and assemble structural variants. Nat Methods. 7(7):485–486.
- Seemann T. 2014. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 30(14):2068–2069.
- Tan XQ, Guo LJ, Zheng W, Tan CH. 2013. Study on chemical components of Fraxinus malacophylla (I). China Pharm. 24(43):4081–4083.