
Abstract
According a recent report by Heidari et al., a mutational screening for candidate pathogenic mitochondrial tRNA (mt-tRNA) mutations were performed in 45 Iranian patients with non-dystrophic myotonia (NDM) and 70 control subjects. Through PCR amplification and direct sequence analysis, nine mt-tRNA mutations were identified: tRNAMet T4454C, tRNATrp A5568G, tRNACys T5794C, tRNAArg A10438T and T10462C, tRNALeu(CUN) A12308G, tRNAThr A15907G, A15924G and G15928A. However, through the database searches and phylogenetic conservation analysis, we noticed that the tRNAThr A15924G, G15928A and tRNALeu(CUN) A12308G mutations should be classified ‘pathogenic’. Thus, the roles of mt-tRNA mutations in clinical expression of NDM needed to be further experimentally addressed.
Introduction
Myotonia was a genetically and heterogeneous disease due to the electrical hyper-excitability of muscle fibers (Morales and Pusch Citation2019). Non-dystrophic myotonia (NDM), a common type of myotonia, had been estimated to be approximately 1 in 100,000 according to a recent report (Snyder et al. Citation2015). To date, the etiology of NDM was still poorly understood. It was well established that high-energy consuming tissues such as muscular and nervous systems were exclusively dependent on the ATP generation by mitochondria; therefore, mitochondrial dysfunction may play an important role in NDM. In fact, mitochondrial proteome consisted of at least 1500 proteins of which 13 were encoded by the mitochondrial DNA (mtDNA) genes (Taylor et al. Citation2003), furthermore, mtDNA encoded 22 tRNAs and two rRNAs. Although the mitochondrial tRNA (mt-tRNA) genes comprised only a small fraction of the mitochondrial genome, however, contributed disproportionately to the etiology of mitochondrial diseases (Chinnery and Hudson Citation2013). To date, over 200 different pathogenic mutations had been mapped to mt-tRNA genes (http://www.mitomap.org/MITOMAP) (Ruiz-Pesini et al. Citation2007), emphasizing the importance of mt-tRNAs for mitochondrial function.
The clinical and molecular diagnosis of mitochondrial diseases may be achieved by mtDNA sequence analysis for known pathogenic mt-tRNA mutations. Nevertheless, a poor genotype to phenotype correlation was very common, as in the case of tRNAMet T4454C (Wang et al. Citation2014) or tRNASer(UCN) T7501C mutation (Ding and Huang Citation2016).
Most recently, Heidari et al. (Citation2020) and colleagues investigated the relationship between mt-tRNA mutations and non-dystrophic myotonia (NDM) in a cohort of 45 Iranian patients and 70 controls. Through genetic amplification of 22 mt-tRNA genes and Sanger sequence analysis, they identified nine mt-tRNA mutations: tRNAMet T4454C, tRNATrp A5568G, tRNACys T5794C, tRNAArg A10438T and T10462C, tRNALeu(CUN) A12308G, tRNAThr A15907G, A15924G and G15928A, and regarded the tRNAArg A10438T as a potential pathogenic mutation for NDM simply because this mutation was statistically significance between NDM and control groups (p < 0.05). However, the pathogenicity of these mt-tRNA mutations remained mysterious. In this study, we examined the genetic susceptibility of these mt-tRNA mutations and further discussed the relationship between these mutations and clinical phenotype.
Materials and methods
Database searches
We carried out the literatures searches for the presence of these nine mt-tRNA mutations via Pubmed Central (https://pubmed.ncbi.nlm.nih.gov) and other public resources (Mitomap database: www.mitomap.org) (Lott et al. Citation2013) with the following keywords: ‘mitochondrial tRNAMet T4454C mutation’; ‘mitochondrial tRNATrp A5568G mutation’; ‘mitochondrial tRNACys T5794C mutation’; ‘mitochondrial tRNAArg A10438T mutation’; ‘mitochondrial tRNAArg T10462C mutation’; ‘mitochondrial tRNALeu(CUN) A12308G mutation’; ‘mitochondrial tRNAThr A15907G mutation’; ‘mitochondrial tRNAThr A15924G mutation; or ‘mitochondrial tRNAThr G15928A mutation’ to identify the case-control studies published to date on the association between these nine mutations and various clinical diseases.
Phylogenetic analysis
To determine the evolutionary conservation of the candidate pathogenic mutations, a phylogenetic approach was performed. Briefly, a total of 15 organisms’ mtDNA sequences were used for this analysis. Moreover, the conservation index (CI) was compared the human mtDNA variations with other 14 species. Notably, the CI > 75% was believed to have functional potential (Levin et al. Citation2013).
Structure analysis
The tRNAMet T4454C, tRNATrp A5568G, tRNACys T5794C, tRNAArg A10438T and T10462C, tRNALeu(CUN) A12308G, tRNAThr A15907G, A15924G and G15928A mutations were individually analyzed using the published secondary structures for the mt-tRNAs with the stem and loop structure (Suzuki et al. Citation2011).
Determining the pathogenicity
We further utilized the updated pathogenicity scoring system to assess these mt-tRNA mutations based on the criteria that generated by Yarham et al. (Citation2011). Notably, if the total scores of the mutation <6, it was classified as ‘neutral polymorphism’, if the scores of the mutation were 7–10, it belonged to ‘possibly pathogenic’, 11–13 points (not including evidence from single fiber, steady-state level, or trans-mitochondrial cybrid studies), it belonged to ‘probably pathogenic’; ≥11 points (including evidence from single fiber, steady-state level or trans-mitochondrial cybrid studies) it was classified as ‘definitely pathogenic’.
Results
Molecular features of nine mt-tRNA mutations
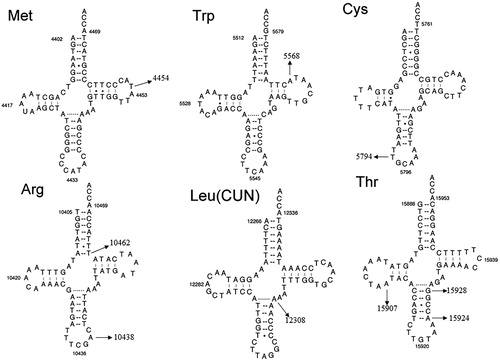
Heidari et al. (Citation2020) identified nine sequence variants in mt-tRNA genes by using PCR and direct sequence. As shown in , these mutations include the T4454C in the T-loop of tRNAMet (position 58), A5568G in the T-stem of tRNATrp (position 62), T5794C in the anticodon stem of tRNACys (position 33), T10462C (position 66) in the acceptor arm and A10438G (position 37) in the anticodon stem of tRNAArg, A12308G in the variable region of tRNALeu(CUN) (position 44), A15907G (position 22) in the D-stem; A15924G (position 39) and G15928A (position 43) in the anticodon stem of tRNAThr (Florentz et al. Citation2003). Of these, the T10462C mutation disrupted the 7 A-66T base-pairing, the A12308G mutation created a new base-pairing (25 A-37T), while the A15924G and G15928A mutations disrupted the highly conserved base-pairings (31 T-39A and 27 C-43G), respectively.
Figure 1. The secondary structure of nine mt-tRNA mutations, arrow indicated the mutation positions.
Analysis of CIs
We performed the phylogenetic conservation analysis for these nine mt-tRNA mutations, as seen and , we found that except for the tRNAArg A10438T, tRNALeu(CUN) A12308G, tRNAThr A15924G and G15928A mutations, other mutations are not conserved, with the CI varied from 21.1% to 82.6%, the lower level of CIs ruled out their roles in clinical expression of mitochondrial diseases.
Figure 2. Sequence alignment of mt-tRNALeu(CUN) gene from 15 different species, arrow indicated the position 44, corresponding to the A12308G mutation.
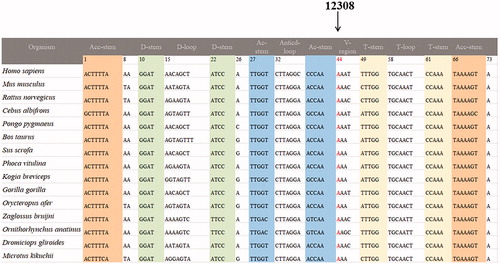
Table 1. Molecular characterization of nine mt-tRNA mutations.
Determining the pathogenicity
According to the updated pathogenicity scoring system (Yarham et al. Citation2011), we noticed that the total scores of mt-tRNALeu(CUN) A12308G, mt-tRNAThr A15924G and G15928A mutations were 15, 13 and 10 points, respectively, belonging to ‘definitely pathogenic’ and ‘possibly pathogenic’. Similarly, the total scores of T4454C, A5568G, T5794C, A10438T, T10462C and A15907G mutations were 4,4,2,4,4 and 2 points, respectively, suggesting that they belong to ‘neutral polymorphisms’ at this stage ().
Table 2. The pathogenicity scoring system for A12308G, A15924G and G15928A mutations.
Discussion
Mitochondrion played a critical role in cellular energy production (Chatterjee et al. Citation2011), in particular, mt-tRNA mutations were being increasingly recognized as important causes for disease, such mutations can result in transcriptional and translational defects and consequently mitochondrial respiratory chain dysfunction (Servidei Citation2003). However, it should be noted that some mutations in mt-tRNA genes cause devastating disease, whereas others had no clinical consequences (McFarland et al. Citation2004).
In the present study, we reassessed the roles of nine mt-tRNA mutations in the phenotypic manifestation of NDM. Among these sequence alternations, the T4454C mutation was located at position 58 in the T-loop of tRNAMet, nucleotide at that position was not well conserved and may not have functional impact on mitochondrial translation (Wang et al. Citation2014). Moreover, the tRNATrp A5568G mutation had been reported to be associated with hearing loss (Jacobs et al. Citation2005), however, no functional analysis was performed in cybrid cells containing this mutation, therefore, the role of A5568G mutation remained controversial. While the homoplasmic T5794C variant occurred at position 33 in the anticodon stem of tRNACys, the A10438T mutation was localized at highly conserved position in the anticodon stem of tRNAArg. Notably, four mutations affected the Watson-Crick base-pairings: the tRNAArg T10462C disrupted the 7 A-66T base-pairing, the tRNALeu(CUN) A12308G created a conserved 25 A-37T base-pairing, whereas the A15924G mutation abolished the 31 T-39A base-pairing, while the G15928A mutation disrupted the 27 C-43G base-pairing ( and ). In addition, by literature searching, we noticed that the tRNALeu(CUN) A12308G mutation had been reported to be associated with CPEO, stroke and breast cancer risk (Pulkes et al. Citation2000; van den Ouweland et al. Citation1992; Covarrubias et al. Citation2008). While the tRNAThr A15924G mutation had been regarded as a risk factor for lethal infantile mitochondrial myopathy (LIMM) and fatal infantile respiratory enzyme deficiency (Brown et al. Citation1992; Yoon et al. Citation1991). Furthermore, the heteroplasmic tRNAThr G15928A mutation was believed to play an important role in multiple sclerosis (MS) and Parkinson’s Disease (PD) (Mayr-Wohlfart et al. Citation1996; 1997; Simon et al. Citation2000). Thus, we supposed that the A12308G, A15924G and G15928A mutations may lead to a failure in mt-tRNAs metabolism, and consequently result the impairment in mitochondrial protein synthesis (Fox Citation2012). In fact, the pathogenicity scoring system indicated that besides the A12308G, A15924G and G15928A mutations, others should be regarded as ‘neutral polymorphisms’ (Yarham et al. Citation2011).
According to these observations, we believed that the possible molecular mechanism underlying the A12308G, A15924G and G15928A mutations in clinical expression of NDM may be as follows, first of all, these mutations altered the secondary structure and affected the steady-state levels of corresponding tRNAs, subsequently, these mutations may lead to the failure in tRNA metabolism such as CCA addition, aminoacylation, or defects in tRNA modifications. As a result, mitochondrial protein synthesis may be affected by these events and the respiratory chain functions impaired, thus, the ATP declined and ROS increased, which led to mitochondrial dysfunction that was involved in the pathogenesis of NDM.
Most recently, several studies had been reported on the associations between mt-tRNA mutations and mitochondrial disorders, as in the cases of T4454C variant and hypertension (Wang et al. Citation2014), the C15891T variant and Leber’s Hereditary Optic Neuropathy (LHON) (Jiang et al. Citation2016), the tRNAPhe C628T variant and hearing loss (Zhu et al. Citation2015). Although we believed that mt-tRNA mutations played important roles in NDM, a call for more carefully reassessment of the dataset seemed necessary. The main limitation of the current study was the lack of functional analysis for these pathogenic mutations; further studies were needed to verify this conclusion.
Acknowledgements
We thanked Dr. Zhaochang Jiang from the Second Affiliated Hospital of Zhejiang University, School of Medicine for critical reading of this manuscript.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
Additional information
Funding
References
- Brown MD, Torroni A, Shoffner JM, Wallace DC. 1992. Mitochondrial tRNAThr mutations and lethal infantile mitochondrial myopathy. Am J Hum Genet. 51(2):446–447.
- Chatterjee A, Dasgupta S, Sidransky D. 2011. Mitochondrial subversion in cancer. Cancer Prev Res (Phila)). 4(5):638–654.
- Chinnery PF, Hudson G. 2013. Mitochondrial genetics. Br Med Bull. 106(1):135–159.
- Covarrubias D, Bai RK, Wong LC, Leal SM. 2008. Mitochondrial DNA variant interactions modify breast cancer risk. J Hum Genet. 53(10):924–928.
- Ding Y, Huang J. 2016. Is mitochondrial tRNA(Ser(UCN)) T7501C mutation associated with cardiovascular disease? Mitochondrial DNA A DNA Mapp Seq Anal. 27(1):205–208.
- Finnila S, Hassinen IE, Majamaa K. 2001. Phylogenetic analysis of mitochondrial DNA in patients with an occipital stroke. Evaluation of mutations by using sequence data on the entire coding region. Mutat Res. 458(1–2):31–39.
- Florentz C, Sohm B, Tryoen-Toth P, Putz J, Sissler M. 2003. Human mitochondrial tRNAs in health and disease. Cell Mol Life Sci. 60(7):1356–1375.
- Fox TD. 2012. Mitochondrial protein synthesis, import, and assembly. Genetics. 192(4):1203–1234.
- Heidari MM, Keshmirshekan A, Bidakhavidi M, Khosravi A, Bandari Z, Khatami M, Nafissi S. 2020. A novel heteroplasmic mutation in mitochondrial tRNAArg gene associated with non-dystrophic myotonias. Acta Neurol Belg. 120(3):573–580.
- Jacobs HT, Hutchin TP, Käppi T, Gillies G, Minkkinen K, Walker J, Thompson K, Rovio AT, Carella M, Melchionda S, et al. 2005. Mitochondrial DNA mutations in patients with postlingual, nonsyndromic hearing impairment. Eur J Hum Genet. 13(1):26–33.
- Jiang Z, Yu J, Xia B, Zhuo G. 2016. Mitochondrial tRNAThr 15891C > G mutation was not associated with Leber’s hereditary optic neuropathy in Han Chinese patients. Mitochondrial DNA A DNA Mapp Seq Anal. 27(2):1564–1566.
- Levin L, Zhidkov I, Gurman Y, Hawlena H, Mishmar D. 2013. Functional recurrent mutations in the human mitochondrial phylogeny: dual roles in evolution and disease. Genome Biol Evol. 5(5):876–890.
- Lott MT, Leipzig JN, Derbeneva O, Xie HM, Chalkia D, Sarmady M, Procaccio V, Wallace DC. 2013. mtDNA variation and analysis using mitomap and mitomaster. Curr Protoc Bioinformatics. 44(123):1.23–1.26.
- Mayr-Wohlfart U, Paulus C, Henneberg A, Rödel G. 1996. Mitochondrial DNA mutations in multiple sclerosis patients with severe optic involvement. Acta Neurol Scand. 94(3):167–171.
- Mayr-Wohlfart U, Rödel G, Henneberg A. 1997. Mitochondrial tRNA(Gln) and tRNA(Thr) gene variants in Parkinson’s disease. Eur J Med Res. 2(3):111–113.
- McFarland R, Elson JL, Taylor RW, Howell N, Turnbull DM. 2004. Assigning pathogenicity to mitochondrial tRNA mutations: when “definitely maybe” is not good enough. Trends Genet. 20(12):591–596.
- Morales F, Pusch M. 2019. An up-to-date overview of the complexity of genotype-phenotype relationships in myotonic channelopathies. Front Neurol. 10:1404.
- Pulkes T, Sweeney MG, Hanna MG. 2000. Increased risk of stroke in patients with the A12308G polymorphism in mitochondria. Lancet. 356 (9247):2068–2069.
- Ruiz-Pesini E, Lott MT, Procaccio V, Poole JC, Brandon MC, Mishmar D, Yi C, Kreuziger J, Baldi P, Wallace DC. 2007. An enhanced MITOMAP with a global mtDNA mutational phylogeny. Nucleic Acids Res. 35(Database issue):D823–828.
- Servidei S. 2003. Mitochondrial encephalomyopathies:gene mutation. Neuromuscul Disord. 13(10):848–853.
- Simon DK, Mayeux R, Marder K, Kowall NW, Beal MF, Johns DR. 2000. Mitochondrial DNA mutations in Complex I and tRNA genes in Parkinson’s disease. Neurology. 54(3):703–709.
- Snyder Y, Donlin-Smith C, Snyder E, Pressman E, Ciafaloni E. 2015. The course and outcome of pregnancy in women with nondystrophic myotonias. Muscle Nerve. 52(6):1013–1015.
- Suzuki T, Nagao A, Suzuki T. 2011. Human mitochondrial tRNAs: biogenesis, function, structural aspects, and diseases. Annu Rev Genet. 45:299–329.
- Taylor SW, Fahy E, Ghosh SS. 2003. Global organellar proteomics. Trends Biotechnol. 21(2):82–88.
- Uusimaa J, Finnilä S, Remes AM, Rantala H, Vainionpää L, Hassinen IE, Majamaa K. 2004. Molecular epidemiology of childhood mitochondrial encephalomyopathies in a Finnish population: sequence analysis of entire mtDNA of 17 children reveals heteroplasmic mutations in tRNAArg, tRNAGlu, and tRNALeu(UUR) genes. Pediatrics. 114(2):443–450.
- van den Ouweland JM, Bruining GJ, Lindhout D, Wit JM, Veldhuyzen BF, Maassen JA. 1992. Mutations in mitochondrial tRNA genes: non-linkage with syndromes of Wolfram and chronic progressive external ophthalmoplegia. Nucleic Acids Res. 20(4):679–682.
- Wang Y, Dong P, Li L, Li X, Wang H, Yang X, Wang S, Li Z, Shang X. 2014. The mitochondrial tRNA(Met) 4454T > C variant may not be associated with essential hypertension in Han Chinese population. Mitochondrial DNA. 25(2):124–125.
- Yarham JW, Al-Dosary M, Blakely EL, Alston CL, Taylor RW, Elson JL, McFarland R. 2011. A comparative analysis approach to determining the pathogenicity of mitochondrial tRNA mutations. Hum Mutat. 32(11):1319–1325.
- Yoon KL, Aprille JR, Ernst SG. 1991. Mitochondrial tRNA(thr) mutation in fatal infantile respiratory enzyme deficiency. Biochem Biophys Res Commun. 176(3):1112–1115.
- Zhu HY, Wang SW, Liu L, Chen R, Wang L, Gong XL, Zhang ML. 2009. Genetic variants in mitochondrial tRNA genes are associated with essential hypertension in a Chinese Han population. Clin Chim Acta. 410(1–2):64–69.
- Zhu Q, Zhou Y, Jin X, Lin X. 2015. The role of mitochondrial tRNAPhe C628T variant in deafness expression. Mitochondrial DNA. 26(1):2–6.
- Zifa E, Theotokis P, Kaminari A, Maridaki H, Leze H, Petsiava E, Mamuris Z, Stathopoulos C. 2008. A novel G3337A mitochondrial ND1 mutation related to cardiomyopathy co-segregates with tRNALeu(CUN) A12308G and tRNAThr C15946T mutations. Mitochondrion. 8(3):229–236.