
Abstract
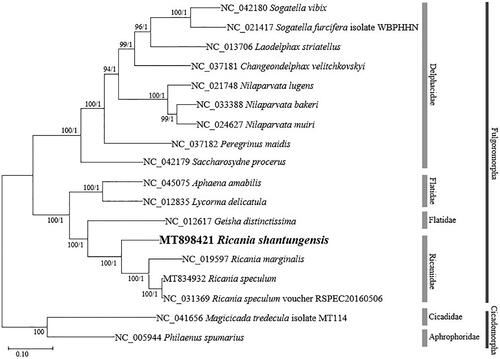
In this study, we have sequenced and annotated the complete mitochondrial genome of Ricania shantungensis (Hemiptera: Ricaniidae) for the first time. The circular mitogenome of R. shantungensis was 15,789 bp, including 13 protein-coding genes, two ribosomal RNA genes, 22 transfer RNAs, and a single control region of 1,363 bp. Its AT ratio was 74.6%. According to the phylogenetic tree, R. shantungensis was clustered with the genus Ricania.
It has been reported that there are 40 genus and 400 species in the family Ricaniidae, including Ricania shantungenesis and the genus Ricania includes about 40 species distributed in Asia including China and India (Xu et al. Citation2006). The original distribution of R. shantungensis is assumed to be Zhejiang and Shandong provinces in China (Chou and Lu Citation1977). It damages host plants by sucking and spawning and causes sooty mold disease. This incurs economic losses for various fruit trees such as blueberries and apples. Recently, it has been continuously spreading throughout Korea and has been detected in 43 cities (Kim et al. Citation2015). In this study, we determined the mitogenome of R. shantungensis.
In August 2018, the sample was collected from Daejeon, Korea (36.114775 N, 127.330402E). Genomic DNA was extracted using DNeasy Blood & Tissue Kit (QIAGEN, Hilden, Germany). The specimen’s genome DNA (Accession number: 973) was deposited in the National Institute of Ecology, Seocheon, Korea. Raw sequences obtained from Illumina HiSeqX (Macrogen Inc., South Korea) were filtered by Trimmomatic 0.33 (Bolger et al. Citation2014) and de novo assembled by Velvet 1.2.10 (Zerbino and Birney Citation2008) and gap sequences were closed with SOAPGapCloser 1.12 (Zhao et al. Citation2011), BWA 0.7.17, and SAMtools 1.9 (Li et al. Citation2009; Li Citation2013). Geneious R11 11.1.5 (Biomatters Ltd, Auckland, New Zealand) was used to annotate the mitochondrial genome of R. shantungensis with referring mitogenomes of R. speculum (NC_031369 and MT834932) and R. marginalis (NC_019597).
The complete mitogenome of R. shantungensis (GenBank accession number: MT898421) was 15,789 bp with 74.6% AT content, which was longer than any other available Ricania mitochondrial genome (R. speculum: 15,729bp). It contained all 39 genes of a standard insect mitochondrial genome, including 13 protein-coding genes, two rRNAs, and 22 tRNAs. The order of the 39 genes was conserved as in all other hemipteran mitogenomes. The single large non-coding control region was also found in the mitogenome, and was 1,363 bp long, the largest of all known Ricania mitogenomes.
We inferred phylogenetic relationships based on multiple sequence alignments of 16 planthopper mitogenomes including two outgroup species, Magicicada tredecula (NC_041656) and Philaenus spumarius (NC_005944) (Song and Liang Citation2013) except variable control regions. Multiple sequence alignment was conducted using MAFFT 7.450 (Katoh and Standley Citation2013). Phylogenetic trees were constructed by maximum likelihood (ML) using MEGA X (Kumar et al. Citation2018) and Bayesian inference (BI) using MrBayes 3.2.6 (Ronquist et al. Citation2012). The tree topology of ML and BI were identical (). According to the tree, R. shantungensis was clustered with two species, R. marginalis and R. speculum, belonging to the genus Ricania. In addition, Ricaniidae was included in a clade with Flatidae. This study will be helpful to understand the phylogenetic relationships of Ricaniidae.
Figure 1. Maximum likelihood (1,000 bootstrap repeats) and Bayesian inference phylogenetic trees of 16 Fulgoromorpha mitochondrial genomes including two outgroup species (Magicicada tredecula and Philaenus spumarius). Numbers on internodes indicate maximum likelihood bootstrap poportions (left) and Bayesian posterior probabilities (right).
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The data that support the findings of this study are openly available in GenBank at https://www.ncbi.nlm.nih.gov/genbank/, accession number [MT898421].
Additional information
Funding
References
- Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 30(15):2114–2120.
- Chou Y, Lu CS. 1977. On the Chinese Ricaniidae with descriptions of eight new species. Acta Entomol Sin. 20:314–322.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30(4):772–780.
- Kim DE, Lee H, Kim MJ, Lee DH. 2015. Predicting the potential habitat, host plants, and geographical distribution of Pochazia shantungensis (Hemiptera: Ricaniidae) in Korea. Korean J Appl Entomol. 54(3):179–189.
- Kumar S, Stecher G, Li M, Knyaz C, Tamura K. 2018. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 35(6):1547–1549.
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. 2009. The sequence alignment/map format and SAMtools. Bioinformatics. 25(16):2078–2079.
- Li H. 2013. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv preprint arXiv:1303.3997.
- Ronquist F, Teslenko M, Van Der Mark P, Ayres DL, Darling A, Höhna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP. 2012. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 61(3):539–542.
- Song N, Liang AP. 2013. A preliminary molecular phylogeny of planthoppers (Hemiptera: Fulgoroidea) based on nuclear and mitochondrial DNA sequences. PLOS One. 8(3):e58400
- Xu CQ, Liang AP, Jiang GM. 2006. The genus Euricania melichar (Hemiptera: Ricaniidae) from China. Raffl Bull Zool. 54(1):1–10.
- Zerbino DR, Birney E. 2008. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18(5):821–829.
- Zhao QY, Wang Y, Kong YM, Luo D, Li X, Hao P. 2011. Optimizing de novo transcriptome assembly from short-read RNA-Seq data: a comparative study. BMC Bioinf. 12(Suppl 14):S2.