
Abstract
Epipedocera atra is a common species of Epipedocera Chevrolat which distributed in South China and some countries in Southeast Asia. The complete mitochondria genome of E. atra was 15,662 bp in length, with 37 genes, including 13 protein-coding genes (PCGs), 22 tRNA genes (tRNAs), and 2 rRNA genes (rRNAs). The nucleotide composition was highly A + T biased, accounting for 70.34% of the whole mitogenome. Phylogenetic analysis indicated that E. atra had a close relationship with Xylotrechus grayii White.
Introduction
Epipedocera atra is a common species of Epipedocera Chevrolat found in some parts of Southeast Asia and South China (Jiang et al. Citation1985; Hua et al. Citation2009). In this study, specimens of E. atra were collected from the Wenjing Village (22°87′N, 109°24′E) of Nanning City (Guangxi Autonomous Region, China) on a corn in middle florescence. The voucher samples were kept at the Guangxi Key Laboratory of Agric-Environment and Agric-Products Safety (Nanning, China), College of Agriculture, Guangxi University, Guangxi, China.
The total genomic DNA was extracted following the modified CTAB DNA extraction protocol and stored at Guangxi Key Laboratory of Agric-Environment and Agric-Products Safety (Nanning, China) with a sample number of SZHT0606G148. Then, a library was constructed and pair-end was sequenced (2*150 bp) with HiSeq (Illumina, San Diego, CA). Approximately 10.40 G of raw data and 10.30 G of clean data were obtained for sequence assembly by SPAdes (Version 3.9) with ‘-k 21,33,55,77 – careful’ option (Bankevich et al. Citation2012). The candidate was obtained by blast alignment of mitochondrial fragments from near-source species. By analyzing the positions of these candidate sequences in fastg graph and combining the adjacency sequence of these sequences in the graph, the complete loop path is obtained. The complete mitochondrial genome was obtained by extracting the sequence from the loop path diagram.
The complete mitochondrial genome of E. atra (GenBank accession number MT740323) revealed the size of 15,662 bp, containing A 38.41%, G 11.49%, T 31.93%, and C 18.17%. It is highly A + T biased, accounting for 70.34%, showing strong AT skew. The E. atra mitogenome contains 13 protein-coding genes (PCGs), 22 tRNAs (tRNA-Leu, tRNA-Gln, tRNA-Met, tRNA-Trp, tRNA-Cys, tRNA-Tyr, tRNA-Second Leu, tRNA-Lys, tRNA-Asp, tRNA-Gly, tRNA-Ala, tRNA-Arg, tRNA-Asn, tRNA-Ser, tRNA-Glu, tRNA-Phe, tRNA-His, tRNA-Thr, tRNA-Pro, tRNA-Second Ser, tRNA-Leu, tRNA-Val), and 2 rRNA (16S rRNA and 12S rRNA). The nucleotide sequence of 13 PCGs of all mitochondrial genes was 11,040 bp in length. The sizes of 2 rRNA genes were1049 bp and 775 bp, respectively. All of the 22 tRNAs, ranging from 62 to71 bp, have a classical cloverleaf structure.
Among these PCGs, 4 genes (nad1, nad2, nad4l, atp8) were with start codon ATT, 4 genes (nad4, atp6, cox1, cox3) have ATG as start codon, 2 genes (cob, cox2) have ATC as start codon, and 2 genes (nad3, nad6) were with start codon ATA. Besides, 8 PCGs (nd2, nd4, nd4l, atp6, cox1, cox2 and cox3) used the stop codon TAA, and the remaining 5 genes (nad1, nad3, nad6, atp8 and cob) were with the stop codon TAG.
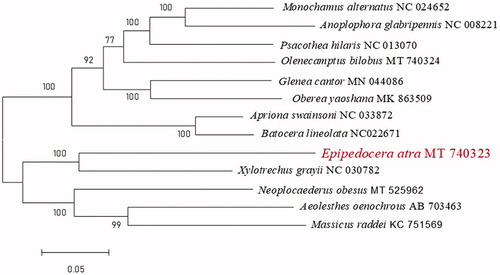
Molecular Evolutionary Genetics Analysis Version 7.0 (MEGA 7.0) was used to make phylogenetic inference among some Cerambycidae species by the maximum-likelihood method with 1000 bootstrap replicates (Kumar et al. Citation2016). Results show that E. atra is sister to Xylotrexhus Chevrolat ().
Figure 1. Maximum-likelihood phylogenetic tree of E. atra and other 12 species in Cerambycidae.
Acknowledgements
We appreciated the sequencing service provided by Huitong biotechnology Co. Ltd (Shenzhen, China).
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
Mitogenome data supporting this study are openly available in GenBank at: https://www.ncbi.nlm.nih.gov/nuccore/MT740323. Associated BioProject, SRA, and BioSample accession numbers are https://www.ncbi.nlm.nih.gov/bioproject/PRJNA663275, https://www.ncbi.nlm.nih.gov/sra/SRX9145190, and SAMN16125319, respectively.
Additional information
Funding
References
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, et al. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. Comput Biol. 19(5):455–477.
- Hua LZ, Nara H, Samuelson GA, Lingafelter SW. 2009. Iconography of Chinese longicorn beetles (1406 species) in color. Guangzhou: Sun Yat-sen University Press.
- Jiang SX, Pu FJ, Hua LZ. 1985. Economic insect fauna of China (Volume III, Coleoptera). Beijing: Science Press (in Chinese).
- Kumar S, Stecher G, Tamura K. 2016. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 33(7):1870–1874.