
Abstract
In this study, the complete mitochondrial genome of Ilisha striatula is presented, and we also discussed its mitochondrial characteristics. The full length of the mitochondrial genome was 16,847 bp, including 13 protein-coding genes (PCGs), 2 ribosomal RNAs, 22 transfer RNA genes, one non-coding control region (CR), and one origin of replication on the light-strand. The total nucleotide composition of the mitochondrial genome was 31.11% A, 24.62% T, 29.10% C, 15.16% G, and AT was 55.73%. The phylogenetic tree based on the ML method supported the placement of I. striatula in a close relationship with I. elongata. These data contribute to a better understanding of the systematics of the Pristigasteridae.
Ilisha striatula (Wongratana Citation1983) is a tropical marine fish classified in the Pristigasteridae of the Clupeiformes, Actinopterygii. It is distributed in the Indian Ocean from Karachi to Kolkata in brackish waters. I. striatula is adapted to depths from 0 to 50 meters and can reach up to 18 cm long. This species inhabits coastal and estuarine waters and is edible (Whitehead Citation1985). Considering its ecological importance, here we describe the complete mitogenome of I. striatula and investigate its phylogenetic relationship within the Pristigasteridae. Phylogenetic studies on the Pristigasteridae are valuable because they provide new information that can improve the propagation and breeding of this species.
The voucher specimen was deposited in the laboratory of Zhejiang Ocean University under accession number 20170826zdl22. Total genomic DNA was extracted from muscle tissue of the specimen following a phenol–chloroform method (Barnett and Larson Citation2012). Using the existing complete mitochondrial genome of I. elongata (AP009141.1) (Lavoué et al. Citation2007) 18 primer pairs were designed, they are: primers 1: For 5′-GCAGGTATCAGGCACAAT-3′ and Rev 5′-TACCTCCTTCAACTAACTCTC-3′; primers 2: For 5′ -GAGAGTTAGTTGAAGGAGGTA-3′ and Rev 5′-GAGTCGTTGTTATTGTGGTT-3′; primers 3: For 5′-AGACACTAACCAACCACAAT-3′ and Rev 5′-AGGCTACTTCGTATGAGATG-3′; primers 4: For 5′-CCACACCATTACGACACA-3′ and Rev 5′-TAGACCGAGGCTTAGTAGTA-3′; primers 5: For 5′-TGAATACACGGACAATGAGA-3′ and Rev 5′-TGAATGCTCGCTGGTTAG-3′; primers 6: For 5′-CCACCATCACAACCATCTT-3′ and Rev 5′-GACCAATACGGCTCAGAC-3′; primers 7: For 5′-CGTATTGGTCACAGCAGTA-3′ and Rev 5′-AGATTAGCGAGCCGATTG-3′; primers 8: For 5′-TCAATCGGCTCGCTAATC-3′ and Rev 5′-CTACTATTCGGTGGTCTGTT-3′; primers 9: For 5′-AACAGACCACCGAATAGTAG-3′ and Rev 5′-AGGCAGATAGCGAGGAAT-3′; primers 10: For 5′-CTCGCTATCTGCCTCTTAC-3′ and Rev 5′-GGTGATGATGATGAGTGTTG-3′; primers 11: For 5′-CCTAACCTGCTGACTCCT-3′ and Rev 5′-CCGCTTGCTACTAATCCTAT-3′; primers 12: For 5′-GCTACGAACGAACTCACA-3′ and Rev 5′-TGGCGAATCAGGCTATTG-3′; primers 13: For 5′-GTAAGCCTCGCATTCCTAA-3′ and Rev 5′-GCCTTGGTTGTGGTGTTA-3′; primers 14: For 5′-TCTAACACCACAACCAAGG-3′ and Rev 5′-CCGAATAAGTCATCCGTAGT-3′; primers 15: For 5′-CATTATACCTCCGACATTGC-3′ and Rev 5′-ATGGCGTATGCGAATAAGA-3′; primers 16: For 5′-TATTCGCATACGCCATTCTA-3′ and Rev 5′-AGCAAGATGTGTAACTGTCA-3′; primers 17: For 5′-TTAACTTCCACCTCTAACTCC-3′ and Rev 5′-CTGTATAGTCAACGCATATCG-3′; primers 18: For 5′-GTATCTATCTCTACTCCACACT-3′ and Rev 5′-TACGCCGAGCATATCAAC-3′. The samples were sequenced using standard Sanger sequencing technology. The complete mitochondrial genome was annotated using Sequin version 15.10 (http://www.ncbi.nlm.nih.gov/Sequin) and tRNA scan-SE version 2.0 (http://trna.ucsc.edu/tRNAscan-SE/) (Todd et al. Citation2016). Transfer RNA genes and their potential cloverleaf structures were identified using tRNAscan-SE (Lowe and Eddy Citation1997). The nucleotide base composition and phylogenetic construction were inferred with MEGA version 6.0 software (Tamura et al. Citation2011). In order to ascertain the phylogenetic status of I. striatula, we used Bayesian inference (BI) and maximum likelihood (ML) optimality criteria to construct a phylogenetic tree based on the 13 protein-coding sequences of 32 mitochondrial genomes. According to the Akaike Information Criteria (AIC), the most suitable nucleotide sequence model was selected through MrModeltest version 2.3 (Yamaoka et al. Citation1978), identifying the most suitable model as GTR + I + G. Two phylogenetic trees were built using the software MrBayes (Ronquist et al. Citation2012) and PhyML (Guindon et al. Citation2010).
The complete mitochondrial genome of I. striatula is a closed circular chromosome composed of 16,847 bp (GenBank number. MK391600.1), consisting of 13 PCGs, 22 tRNA genes, 2 rRNA genes, 1 replication origin (OL), and a control region (CR). The overall base composition is 31.11% A, 24.62% T, 29.10% C, and 15.16% G. Twelve PCGs, 14 tRNA genes, and two rRNA genes are located on the heavy strand, while one PCG (ND6) and eight tRNA genes (tRNAGln, RNAAla, tRNAAsn, tRNACys, tRNATyr, tRNASer, tRNAGlu, and tRNAPro) on the light strand. All the PCGs use the initiation codon ATG except COI (GTG), which is common in teleost (Li et al. Citation2014; Hui and Xian Citation2015). Most of the protein-coding genes (PCGs) terminate with TAA or TAG as the stop codon, except COII, ND4, and Cytb, which have an incomplete stop codon T–. A total of 52 base pairs in 11 intergenic spacers are found ranging from 1 to 30 bp in length. The lengths of 22 tRNAs distributed on the H and L strands are between 66 and 74 bp. Except for tRNASer (GCT), lacking a DHU stem among the 22 tRNAs, the remaining tRNAs form a conserved clover secondary structure. The lengths of the two rRNA genes are 959 bp (12S rRNA) and 1692 bp (16S rRNA), respectively, and are located between tRNAPhe and tRNALeu and separated by the tRNAVal gene. The length of the control region was 1192 bp, located between tRNAPhe and tRNAPro.
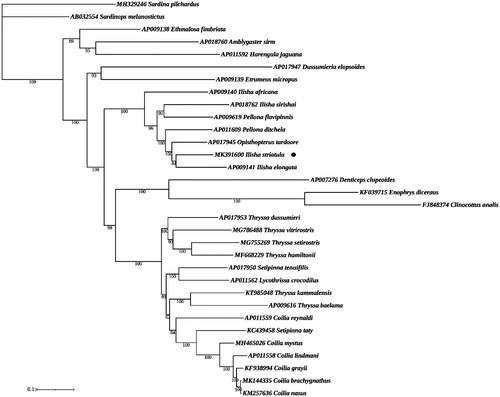
The phylogenetic analyses showed that the BI and ML trees are congruent in topology, with the ML shown here (). I. striatula was closely related to I. elongata (). A similar phylogenetic relationship was inferred (Lavoué et al. Citation2007), which also provided essential and important DNA molecular data for further phylogenetic and evolutionary analysis for species classified in the Clupeiformes.
Figure 1. Phylogenetic analysis based on the nucleotide sequences of the 13 PCGs in the mitogenome. Maximum Likelihood analyses (bootstrap support with 1000 replications) are shown next to the nodes. The number before the species name is the GenBank accession number. The genome sequence in this study is labeled with a black dot.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The data that support the findings of this study are openly available in “NCBI” at https://www.ncbi.nlm.nih.gov/nuccore/MK391600, GenBank number: MK391600.1.
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article.
Additional information
Funding
References
- Barnett R, Larson G. 2012. A phenol-chloroform protocol for extracting DNA from ancient samples. Methods Mol Biol. 840:13–19.
- Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. 2010. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0%. J Syst Biol. 59(3):307–321.
- Hui Z, Xian W. 2015. The complete mitochondrial genome of the larvae Japanese anchovy Engraulis japonicus (Clupeiformes, Engraulidae). Mitochondrial DNA. 26:935–936.
- Lavoué S, Miya M, Saitoh K, Ishiguro NB, Nishida MJMP. 2007. Phylogenetic relationships among anchovies, sardines, herrings and their relatives (Clupeiformes), inferred from whole mitogenome sequences. Mol Phylogenet Evol. 43(3):1096–1105.
- Li C, Lu CY, Li JT, Cheng L, Zheng XH, Sun XW. 2014. Complete mitochondrial genome of Amur sturgeon (Acipenser schrenckii). Mitochondrial DNA. 25(4):282–283.
- Lowe TM, Eddy SR. 1997. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25(5):955–964.
- Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Höhna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP. 2012. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 61(3):539–542.
- Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. 2011. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 28(10):2731–2739.
- Todd M, Lowe, Patricia P. 2016. tRNAscan-SE on-line: integrating search and context for analysis of transfer RNA genes. Nucleic Acid Res. 44(W1):W54–W57.
- Whitehead PJP. 1985. FAO species catalogue: vol. 7. Clupeoid fishes of the world (Suborder Clupeoidei): an annotated and illustrated catalogue of the herrings, sardines, pilchards, sprats, chads, anchovies and wolf-herrings: 1. Chirocentridae, Clupeidae and Pristigasteridae. London: British Museum.
- Wongratana T. 1983. Diagnoses of 24 new species and proposal of a new name for a species of Indo-Pacific Clupeoid fishes. Jpn J Ichthyol. 29(4):385–407.
- Yamaoka K, Nakagawa T, Uno T. 1978. Application of Akaike’s Information Criterion (AIC) in the evaluation of linear pharmacokinetic equations. J Pharmacokinet Biopharm. 6(2):165–175.