
Abstract
The genus Lasius is a conspicuous and popular genus of ants found in the Holarctic regions. We have completed the mitochondrial genome of Lasius spathepus as the first mitochondrial genome of Lasius. The mitochondrial genome is 18,951 bp long, which is the sixth longest ant mitochondrial genome known to science. It contains 13 protein-coding genes, 2 ribosomal RNAs, 22 transfer RNAs, and a control region in a gene order shared with other species of subfamily Formicinae. The control region is 2,147 bp long, longest of all ants. Phylogenetic analysis shows L. spathepus groups with Nylanderia flavipes of the same tribe Lasiini.
Genus Lasius is a common, diverse, and conspicuous group of ants found in the Holarctic regions (Wilson Citation1955). They are not only one of the most abundant and dominant ants of such regions but are also remarkable in showing fascinating behaviors, such as social parasitism or fungiculture (Maruyama et al. Citation2008). These ants therefore have drawn much attention of European and North American entomologists, resulting in various researches of the biology and taxonomy of these ants, eventually becoming the very foundations of many parts of modern Myrmecology (Wilson Citation1955). Ironically, the genus has not received much notice in the aspects of mitochondrial genomes. As the first mitochondrial genome in genus Lasius and the second in tribe Lasiini, we completed the mitochondrial genome of Lasius spathepus, a socially parasitic species of subgenus Dendrolasius common in far east Asia.
The ants were collected from a well-established colony found nesting under a willow tree in the campus of Korea University, Seoul, Republic of Korea (37°35'03.2″N 127°01'37.8″E). Total DNA was extracted from worker ants using DNeasy Blood & Tissue Kit (QIAGEN, Hilden, Germany). Sequencing library was constructed using Illumina TruSeq Nano DNA Library Preparation Kit (Illumina, San Diego, CA) following manufacturer’s recommendations with around 350-bp DNA fragments. 5.97 Gbp raw sequences obtained from Illumina HiSeqX at Macrogen Inc., Korea, were filtered by Trimmomatic v0.33 (Bolger et al. Citation2014), de novo assembled and confirmed by Velvet v1.2.10 (Zerbino and Birney Citation2008), SOAPGapCloser v1.12 (Zhao et al. Citation2011), BWA v0.7.17 (Li et al. Citation2009), and SAMtools v1.9 (Song and Liang Citation2013) under the environment of Genome Information System (GeIS; http://geis.infoboss.co.kr; Park et al., in preparation). Geneious R11 v11.1.5 (Biomatters Ltd, Auckland, New Zealand) was used for annotating the mitochondrial genome based on alignments with other ant mitogenomes and MITOS (Bernt et al. Citation2013) was used to double-check annotated genes. DNA sample and specimen (95% ethanol) were deposited in InfoBoss Cyber Herbarium (IN; http://herbarium.infoboss.co.kr/; Contact person: Suhyeon Park, [email protected]) under the voucher number, KFDS00110.
The new mitochondrial genome of L. spathepus (GenBank accession: MW074965) is 18,951 bp long, 17.5% in GC ratio, and contains 37 standard genes of insect mitochondrial genomes, which includes 13 protein-coding genes, 2 rRNA genes, and 22 tRNA genes (Cameron Citation2014). Its large genome size is outmatched only by five ant species, four leaf-cutter ant species from genus Atta and Acromyrmex (Atta sexdens; MF591717; 19,748 bp, Atta texana; MF417380; 19,709 bp, Acromyrmex echinator; MK861063; 19,550 bp, Atta opaciceps; KY950643; 19,257bp; Barbosa et al. Citation2019) and a collared ant species from genus Aphaenogaster (Aphaenogaster famelica NC_049859: 19,464 bp; Park et al. Citation2020a). The AT-rich control region of L. spathepus mitogenome presented a whopping length of 2,147 bp, longer than any other ants mitogenomes since the extra bases of other ant mitochondrial genomes were dispersed in multiple intergenic regions while those of L. spathepus mitogenome were concentrated in the control region. The order of the 37 genes was identical to that shared by many other formicine species, which is considered the ancestral gene order of both subfamily Formicinae and family Formicidae (Babbucci et al. Citation2014; Vieira and Prosdocimi Citation2019). Translocation of trnI and inversion of trnP identified in Nylanderia flavipes (NC_049861; Park et al. Citation2020b) were not present in L. spathepus, thus are not traits shared by the whole tribe but are derived features for N. flavipes.
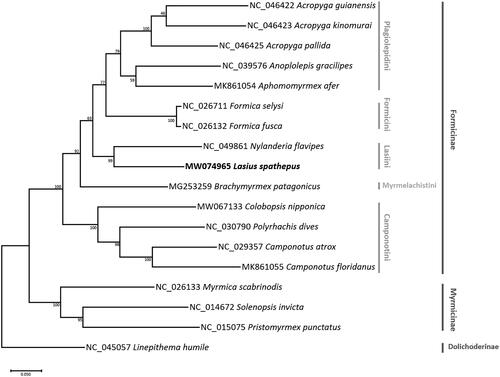
Thirteen PCGs and two rRNA genes from 14 Formicinae ants including L. spathepus and 4 outgroup ant species were aligned independently using MAFFT v7.450 (Katoh and Standley Citation2013) and were concatenated for phylogenetic purposes. Maximum-likelihood (ML) tree was drawn using MEGA X (Kumar et al. Citation2018) in which a heuristic search was used with nearest-neighbor interchange (NNI) branch swapping, Tamura–Nei model, and uniform rates among sites. In the phylogenetic results, L. spathepus grouped with N. flavipes, resulting as a monophyletic branch of tribe Lasiini (). Tribe Lasiini was sister to the clade of tribe Formicini and Plagiolepidini (). This tribal topology was congruent to previous phylogenomic analysis (Ward et al. Citation2016), with the exception of tribe Camponotini, as it was sister to all other formicine tribes (), not nested in the subfamily as in the phylogenomic tree (Ward et al. Citation2016).
Figure 1. Maximum-likelihood (bootstrap repeat: 10,000) phylogenetic tree of 14 Formicinae ant mitochondrial genomes. The numbers above branches indicate bootstrap support values of the maximum likelihood tree. Tribe and subfamily names are presented on the right side of the tree with gray and dark gray colors, respectively.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
Mitochondrial genome sequence can be accessed via accession number MW074965 in GenBank of NCBI at https://www.ncbi.nlm.nih.gov. The associated BioProject, SRA, and Bio-Sample numbers are PRJNA667958, SAMN16392959, and SRR12791243, respectively.
Additional information
Funding
References
- Babbucci M, Basso A, Scupola A, Patarnello T, Negrisolo E. 2014. Is it an ant or a butterfly? Convergent evolution in the mitochondrial gene order of Hymenoptera and Lepidoptera. Genome Biol Evol. 6(12):3326–3343.
- Barbosa JT, Barbosa MS, Morais S, Santana AE, Almeida C. 2019. Mitochondrial genomes of genus Atta (Formicidae: Myrmicinae) reveal high gene organization and giant intergenic spacers. Genet Mol Biol. 42(4), e20180055.
- Bernt M, Donath A, Jühling F, Externbrink F, Florentz C, Fritzsch G, Pütz J, Middendorf M, Stadler PF. 2013. MITOS: improved de novo metazoan mitochondrial genome annotation. Mol Phylogenet Evol. 69(2):313–319.
- Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 30(15):2114–2120.
- Cameron SL. 2014. Insect mitochondrial genomics: implications for evolution and phylogeny. Annu Rev Entomol. 59:95–117.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30(4):772–780.
- Kumar S, Stecher G, Li M, Knyaz C, Tamura K. 2018. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 35(6):1547–1549.
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. 2009. The sequence alignment/map format and SAMtools. Bioinformatics. 25(16):2078–2079.
- Maruyama M, Steiner FM, Stauffer C, Akino T, Crozier RH, Schlick-Steiner BC. 2008. A DNA and morphology based phylogenetic framework of the ant genus Lasius with hypotheses for the evolution of social parasitism and fungiculture. BMC Evol Biol. 8(1):237.
- Park J, Xi H, Park J. 2020a. The complete mitochondrial genome of Aphaenogaster famelica (Smith, 1874) (Hymenoptera: Formicidae)). Mitochondrial DNA Part B. 5(1):492–494.
- Park J, Xi H, Park J. 2020b. The complete mitochondrial genome of Nylanderia flavipes (Smith, 1874)(Hymenoptera: Formicidae). Mitochondrial DNA Part B. 5(1):420–421.
- Song N, Liang A-P. 2013. A preliminary molecular phylogeny of planthoppers (Hemiptera: Fulgoroidea) based on nuclear and mitochondrial DNA sequences. PLOS One. 8(3):e58400.
- Vieira GA, Prosdocimi F. 2019. Accessible molecular phylogenomics at no cost: obtaining 14 new mitogenomes for the ant subfamily Pseudomyrmecinae from public data. PeerJ. 7:e6271.
- Ward PS, Blaimer BB, Fisher BL. 2016. A revised phylogenetic classification of the ant subfamily Formicinae (Hymenoptera: Formicidae), with resurrection of the genera Colobopsis and Dinomyrmex. Zootaxa. 4072(3):343–357.
- Wilson EO. 1955. A monographic revision of the ant genus Lasius. Bull Museum Comparative Zool. 113:1–201.
- Zerbino DR, Birney E. 2008. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18(5):821–829.
- Zhao QY, Wang Y, Kong YM, Luo D, Li X, Hao P. 2011. Optimizing de novo transcriptome assembly from short-read RNA-Seq data: a comparative study. BMC Bioinformatics. 12(Suppl 14):S2.