
Abstract
Mitochondrial DNA is maternally inherited and is shown to affect nuclear–cytoplasmic interactions in potato. Analyzing the mitogenome helps understand the evolutionary relationships and improve breeding programs in potato. We report complete mitogenome sequences from a panel of 13 potato accessions of various taxa. Each mitogenome has three independent circular molecules, except one of the S. bukasovii sample BUK2, which has a single circular molecule. Each mitogenome code for 37 non-redundant protein-coding genes, three rRNAs, 20 tRNAs, and 19 hypothetical open reading frames. Phylogenetic analysis reveals congruency between plastome and mitogenome phylogeny.
Keywords:
Introduction
Plant mitochondrial genomes, mitogenomes, are larger and more complex compared to those of other eukaryotic organisms (Varré et al. Citation2019). The structure and organization of potato mitogenomes include circular and linear conformations with sub-genomic molecules generated by recombination events at repeat regions (Cho et al. Citation2017; Varré et al. Citation2019). In addition to functions in common with other organisms’ mitochondria, such as respiration, metabolism, and programmed cell death, plant mitochondria also have a function in male fertility (Kozik et al. Citation2019). Mitochondrial genomes show divergent evolution that affects nuclear–cytoplasmic interactions (Grun Citation1990). These interactions are important in breeding when intercrossing between related Solanum species, which are riddled with incompatibles. However, complete mitogenome sequences have only been reported for two potato species, S. tuberosum and S. commersonii, to date (Cho et al. Citation2017, Citation2018; Varré et al. Citation2019). Improving genetic resources helps to better understand the genetic diversity within potato. The current study examines complete mitochondrial genome sequences from 13 accessions of diverse tuber-bearing Solanum taxa (Achakkagari et al. Citation2020).
Materials and methods
DNA extraction and sequencing
Total genomic DNA was extracted from the leaves of 13 potato accessions (Solanum stenotomum subsp. goniocalyx (GON1) (CIP Citation2021a), S. stenotomum subsp. goniocalyx (GON2) (CIP Citation2021b), S. phureja (PHU) (CIP Citation2021c), S. xajanhuiri (AJH) (CIP Citation2021d), S. stenotomum subsp. stenotomum (STN) (CIP Citation2021e), S. bukasovii (BUK1) (CIP Citation2021f), S. bukasovii (BUK2) (CIP Citation2021g), S. tuberosum subsp. andigena (ADG1) (CIP Citation2021h), S. tuberosum subsp. andigena (ADG2) (CIP Citation2021i), S. curtilobum (CUR) (CIP Citation2021j), S. juzepczukii (JUZ) (CIP Citation2021k), S. chaucha (CHA) (CIP Citation2021l), and S. tuberosum subsp. tuberosum (TBR) (CIP Citation2021m)) grown at the International Potato Center (CIP) in Lima, Peru as described (Kyriakidou et al. Citation2020), and sequenced using Illumina PE technology, except BUK2, which was sequenced with 10X Genomics’ GemCode technology (https://www.10xgenomics.com/).
Mitogenome assembly and annotation
The raw reads obtained from Illumina sequencing were initially processed using Trimmomatic v0.39 (Bolger et al. Citation2014). The parameters used for the Trimmomatic are ILLUMINACLIP: TruSeq3-PE. Fa: 2:30:10 LEADING:3 TRAILING:3 SLIDINGWINDOW: 4:15 MINLEN:60. The reads of BUK2 obtained from the 10X Genomics’ GemCode technology were trimmed using a python script ‘filter_10xReads.py’ (Davis Citation2021). Potato mitochondrial genome sequences available from the GenBank were downloaded and concatenated to create a reference set (GenBank accession no: MF989953.1, MF989954.1, MF989955.1, MF989956.1, MF989957.1, MF989958.1, MF989959.1, MF989960.1, MF989961.1, MN1044801.1, MN104802.1, MN104803.1, MN114537.1, MN114537.2, and MN114537.3) (Cho et al. Citation2017; Varré et al. Citation2019). The filtered reads of each mitogenome from the CIP panel of potato species were mapped against this reference set. The mapped reads were extracted and used in an initial assembly with GetOrganelle (Jin et al. Citation2019). The longest contigs in the assembly were selected based on Blast searches against the reference set and then used as seed contigs for further assembly (Johnson et al. Citation2008). The seed contigs were extended iteratively to get a complete mitogenome sequence using NOVOPlasty v4.2 (Dierckxsens et al. Citation2016). Filtered WGS reads were used for the extension to retain unique mitochondrial sequences. For BUK2, an initial assembly was carried out using NOVOPlasty v4.2 with filtered reads (Dierckxsens et al. Citation2016). Then, filtered reads of BUK2 were mapped to the reference set along with NOVOPlasty-assembled contigs. The fastq files suited for supernova were generated from the mapped reads using a python script ‘regen_10xReads.py’ (Davis Citation2021), and executed supernova with –accept-extreme-coverage option (Weisenfeld et al. Citation2017). The supernova-assembled scaffolds along with the NOVOPlasty-assembled contigs were run through tigmint, arcs, and LINKS to correct misassemblies and to further assemble into scaffolds (Warren et al. Citation2015; Yeo et al. Citation2017; Jackman et al. Citation2018). The assembled sequences were checked for structural errors using NucBreak (Khelik et al. Citation2020), and then annotated using GeSeq with the reference set of mitogenomes (Tillich et al. Citation2017). The annotations were manually examined and curated using BLAST searches (Johnson et al. Citation2008).
Phylogeny
The coding sequences of common protein-coding genes (atp4, atp8, atp9, ccmB, ccmC, ccmFc, ccmFn, cob, cox1, cox2, cox3, matR, mttB, nad3, nad4L, nad6, nad7, nad9, rpl10, rpl2, rpl5, rps12, rps13, rps19, rps3, rps4, sdh3, sdh4) were extracted and concatenated from each mitogenome, and used to construct a phylogenetic tree (Varré et al. Citation2019). The reference mitogenomes used in this study were also added to the phylogeny. Sequences were then aligned using MAFFT v7.471 (Katoh et al. Citation2002) and a maximum parsimony phylogenetic tree was constructed using MEGA-X (Kumar et al. Citation2018) with 1000 bootstrap replicates.
Results and discussion
Mitogenome assembly
The mitogenome of each of the 13 CIP potato accessions was assembled and the results show that they range in size from 429,483 bp to 478,227 bp (GenBank accession MW122949–MW122985). Each mitogenome was assembled into three independent circular molecules, except BUK2, which was assembled into a single circular molecule (). Molecule 1 ranges from 49,155 bp to 49,302 bp between 12 mitogenomes. Similarly, molecule 2 and 3 range from 111,694 bp to 113,545 bp and 284,372 bp to 316,322 bp, respectively. BUK2 was excluded since it does not have a discernible molecule 1, 2, and 3. Plant mitochondria are very complex, and recombination events lead to the presence of alternative arrangements. Previous studies have also reported similar mitogenome conformations in potato species. Five circular DNA molecules were reported in S. tuberosum (Cho et al. Citation2017), two circular DNA molecules in S. commersonii (Cho et al. Citation2018), and recently two circular and one linear conformation were observed in two S. tuberosum cultivars (Varré et al. Citation2019). Numerous direct/inverted repeat sequences are present in each mitogenome. A few repeat sequences are larger than 1000 bp and are conserved between the mitogenomes. The largest repeat sequence ranges from 11,309 bp to 11,916 bp, and is present in all of the 13 mitogenomes (). The smallest repeat sequence is 1589 bp, and is present in all of the mitogenomes, except BUK2. The repeat R2 is present only in ADG1, ADG2, and CHA. Similarly, the R4 repeat is only present in GON1, GON2, PHU, STN, BUK1, and CUR, mitogenomes where the repeat structure is generally the same. The repeat structure in ADG2 and CHA, AJH and JUZ is also generally the same. A previous study reported that five repeats, which are larger than 1000 bp are present in two of the S. tuberosum cultivars (Varré et al. Citation2019). The repeats R1, R3, R4, and R5 mentioned below were found in these two cultivars as well. However, a 1208 bp repeat present in these two cultivars is missing in all the 13 CIP mitogenomes in the present study.
Table 1. Conformation and size of the observed molecules present in the mitogenomes of 13 potato cultivars.
Table 2. Repeats and their size in each mitogenome.
Mitogenome annotation
The 13 mitogenomes were annotated to determine their gene content and organization. Each encode 37 non-redundant protein-coding genes, three rRNAs, and 20 tRNAs. In addition, each mitogenome has 19 non-redundant hypothetical open reading frames. The majority of mitogenomes had internal stop codons in orf111 (GON1, GON2, PHU, STN, BUK1, ADG1, ADG2, JUZ, and CUR). Similarly, orf140 in BUK2 contained internal stop codons, whereas orf123 in the TBR mitogenome was truncated at the 5′ end. A pseudogene rps14 and a truncated copy of the cob gene are present in all the genomes, except BUK2. The arrangement of Ψrps14-Ψcob was previously observed in S. tuberosum accessions (Varré et al. Citation2019). Due to the presence of repeats that contained duplicated genes, the AJH, ADG1, ADG2, JUZ, CHA, and TBR mitogenomes have a higher number of total genes compared to the rest; however, the unique set of genes remains the same among all of them. The genes cox2, rpl16, rps19, rps3, and orf102 are duplicated in AJH, JUZ, CHA, ADG2, and TBR mitogenomes due to the presence of the R3 repeat sequence. This duplication was also reported in two S. tuberosum cultivars (Varré et al. Citation2019). Similarly, orf131 is duplicated in ADG1, ADG2, and CHA due to the presence of the R2 repeat sequence. A ribosomal protein rps1 is duplicated only in BUK2.
Phylogenetic analysis
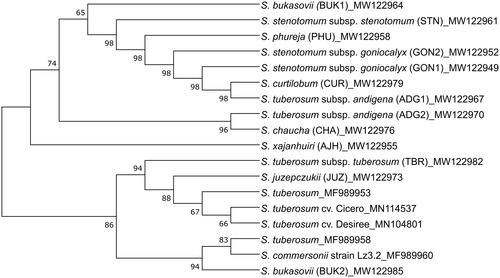
Mitochondrial DNA is maternally inherited and can be used to accurately identify evolutionary relationships. Generating nuclear phylogeny along with organelle phylogeny can be an effective help in understanding the history of hybridization (Achakkagari et al. Citation2020). From the phylogenetic reconstruction, it was observed that GON1, GON2, PHU, STN, BUK1, ADG1, and CUR accessions are grouped together with no significant genetic variation (). A previous study reported a similar classification for the S. stenotomum subsp. goniocalyx, S. phureja, and S. stenotomum subsp. stenotomum species based on six mitogenome markers (Bonen et al. Citation2007). Similarly, ADG2 and CHA mitogenomes are grouped together. The nuclear and plastome phylogeny of these 13 accessions was previously determined (Achakkagari et al. Citation2020; Kyriakidou et al. Citation2020), and a similar type of grouping was observed in these potato accessions. Also, a similar grouping of AJH, JUZ, and TBR accessions was observed in the plastome phylogeny. It is interesting to see all the S. tuberosum species in one clade, except for ADG1 and ADG2. The BUK2, which was previously observed to have close relations with wild species, is grouped with S. commersonii here. The two accessions of S. bukasovii (BUK1 and BUK2) are phylogenetically distant from each other. Similar results were observed in a previous study, and it is likely due to the collection of this accession as a natural population (Achakkagari et al. Citation2020). The reference species are confined to a single clade in this phylogeny; however, the accessions from our panel are widespread across the phylogeny, representing a wider variety of Solanum taxa.
Figure 1. The Phylogenetic tree of 18 mitogenomes from a range of Solanum species (cultivated and wild). The GenBank accession of each mitogenome is provided in the figure.
Conclusions
The mitogenome of 13 potato accessions from a selected panel was assembled and annotated. Three independent circular conformations were observed in all the accessions, except BUK2. The repeat structure in these mitogenomes is interesting and varies from accession to accession. The core genes are similar in all the accessions; however, the AJH, ADG1, ADG2, JUZ, CHA, and TBR mitogenomes have duplicated genes resulting from the repeat sequences. Finally, the phylogenetic relationships between these species were determined. The clustering of these species is mostly in agreement with the previous studies. This is the first study that reports complete mitogenome sequences from a panel that represents seven species, nine taxa, and two wild relatives based on Hawkes taxonomy (Hawkes Citation1990). The results of this study will greatly improve the genetic resources of potato mitogenome. It will also be useful in future comparative studies to better understand the evolutionary relationships in potato species.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The genome sequence data that support the findings of this study are openly available in GenBank of NCBI at https://www.ncbi.nlm.nih.gov/nuccore/ under the accession numbers MW122949–MW122985. The associated BioProject number is PRJNA556263, SRA accession numbers are SRR10248510–SRR10248515, SRR10244436–SRR10244441, and BioSample numbers are SAMN12684886–SAMN12684896, SAMN12345900.
References
- Achakkagari SR, Kyriakidou M, Tai HH, Anglin NL, Ellis D, Strömvik MV. 2020. Complete plastome assemblies from a panel of 13 diverse potato taxa. PLOS One. 15(10):e0240124.
- Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 30(15):2114–2120.
- Bonen L, Scotti NS, Cozzolino SC, Cardi TC. 2007. Mitochondrial DNA variation in cultivated and wild potato species (Solanum spp.) Contribution No. 87 from the Institute of Plant Genetics, Research Division of Portici. Genome. 50(8):706–713.
- Cho K-S, Cho J-H, Im J-S, Choi J-G, Park Y-E, Hong S-Y, Kwon M, Kang J-H, Park T-H. 2017. The complete mitochondrial genome sequences of potato (Solanum tuberosum L., Solanaceae). Mitochondrial DNA Part B. 2(2):781–782.
- Cho K-S, Cho J-H, Im J-S, Choi J-G, Park Y-E, Hong S-Y, Park T-H. 2018. Mitochondrial genome sequence of tuber-bearing wild potato, Solanum commersonii Dunal. Mitochondrial DNA Part B. 3(1):198–199.
- CIP. 2021a. CIP 702472. Solanum stenotomum subsp. goniocalyx (GON1).
- CIP. 2021b. CIP 704393. Solanum stenotomum subsp. goniocalyx (GON2).
- CIP. 2021c. CIP 703654. Solanum phureja (PHU).
- CIP. 2021d. CIP 703810. Solanum xajanhuiri (AJH).
- CIP. 2021e. CIP 705834. S. stenotomum subsp. stenotomum (STN).
- CIP. 2021f. CIP 761748. Solanum bukasovii (BUK1).
- CIP. 2021g. CIP 761748. Solanum bukasovii (BUK2).
- CIP. 2021h. CIP 700921. Solanum tuberosum subsp. andigena (ADG1).
- CIP. 2021i. CIP 702853. Solanum tuberosum subsp. andigena (ADG2).
- CIP. 2021j. CIP 702937. Solanum curtilobum (CUR).
- CIP. 2021k. CIP 706050. Solanum juzepczukii (JUZ).
- CIP. 2021l. CIP 707129. Solanum chaucha (CHA).
- CIP. 2021m. CIP 705053. Solanum tuberosum subsp. tuberosum (TBR).
- Davis U. 2021. Bioinformatics core. https://github.com/ucdavis-bioinformatics/proc10xG.
- Dierckxsens N, Mardulyn P, Smits G. 2016. NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 45(4):e18.
- Grun P. 1990. The evolution of cultivated potatoes. Econ Bot. 44(S3):39–55.
- Hawkes JG. 1990. The potato: evolution, biodiversity and genetic resources. London: Belhaven Press; p. 259.
- Jackman SD, Coombe L, Chu J, Warren RL, Vandervalk BP, Yeo S, Xue Z, Mohamadi H, Bohlmann J, Jones SJM, et al. 2018. Tigmint: correcting assembly errors using linked reads from large molecules. BMC Bioinformatics. 19(1):393.
- Jin J-J, Yu W-B, Yang J-B, Song Y, dePamphilis CW, Yi T-S, Li D-Z. 2019. GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. bioRxiv. 256479.
- Johnson M, Zaretskaya I, Raytselis Y, Merezhuk Y, McGinnis S, Madden TL. 2008. NCBI BLAST: a better web interface. Nucleic Acids Res. 36(Web Server issue):W5–W9.
- Katoh K, Misawa K, Kuma K, Miyata T. 2002. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30(14):3059–3066.
- Khelik K, Sandve GK, Nederbragt AJ, Rognes T. 2020. NucBreak: location of structural errors in a genome assembly by using paired-end Illumina reads. BMC Bioinformatics. 21(1):66.
- Kozik A, Rowan BA, Lavelle D, Berke L, Schranz ME, Michelmore RW, Christensen AC. 2019. The alternative reality of plant mitochondrial DNA: one ring does not rule them all. PLOS Genet. 15(8):e1008373.
- Kumar S, Stecher G, Li M, Knyaz C, Tamura K. 2018. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 35(6):1547–1549.
- Kyriakidou M, Achakkagari SR, Gálvez López JH, Zhu X, Tang CY, Tai HH, Anglin NL, Ellis D, Strömvik MV. 2020. Structural genome analysis in cultivated potato taxa. Theor Appl Genet. 133(3):951–966.
- Tillich M, Lehwark P, Pellizzer T, Ulbricht-Jones ES, Fischer A, Bock R, Greiner S. 2017. GeSeq- versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 45(W1):W6–W11.
- Varré J-S, D'Agostino N, Touzet P, Gallina S, Tamburino R, Cantarella C, Ubrig E, Cardi T, Drouard L, Gualberto JM, et al. 2019. Complete sequence, multichromosomal architecture and transcriptome analysis of the Solanum tuberosum mitochondrial genome. Int J Mol Sci. 20(19):4788.
- Warren RL, Yang C, Vandervalk BP, Behsaz B, Lagman A, Jones SJM, Birol I. 2015. LINKS: scalable, alignment-free scaffolding of draft genomes with long reads. Gigascience. 4(1):35.
- Weisenfeld NI, Kumar V, Shah P, Church DM, Jaffe DB. 2017. Direct determination of diploid genome sequences. Genome Res. 27(5):757–767.
- Yeo S, Coombe L, Chu J, Warren RL, Birol I. 2017. ARCS: assembly roundup by chromium scaffolding. bioRxiv. 100750.