
Abstract
Strobilanthes bantonensis Lindau belongs to the family Acanthaceae. It is an antiviral herb that can be used to prevent Influenza virus infections in the border areas between China and Vietnam. Local people call it ‘Purple Ban-lan-gen’ because its root is very similar to that of Strobilanthes cusia (Nees) Kuntze, which is called ‘Southern Ban-lan-gen’ and is listed in Chinese Pharmacopeia. The two species have been used interchangeably locally. However, their pharmacological equivalence has caused concern for years. We have sequenced the chloroplast genome of S. cusia previously. In this study, we sequenced the complete chloroplast genome sequence of S. bantonensis to preform in-depth comparative genetic analysis of the two Strobilanthes species. The chloroplast genome of S. bantonensis is a circular DNA molecule with a total length of 144,591 bp and encodes 84 protein-coding, 8 ribosomes, and 37 transfer RNA genes. The chloroplast genome has a conservative quadripartite structure, including a large single-copy (LSC) region, a small single-copy (SSC) region, and a pair of inverted repeat (IR) regions, with lengths of 92,068 bp, 17,767 bp, and 17,378 bp, respectively. Phylogenetic analysis confirmed that S. bantonensis is closely related to the S. cusia. Compared with other species from Acanthaceae, S. bantonensis has a significantly shortened IR region, suggesting the occurrence of IR contraction events. This study will help future taxonomic, evolutionary, phylogenetic, and bioprospecting studies of the sizeable Strobilanthes genus, which contains over 400 species.
1. Introduction
Strobilanthes bantonensis Lindau is a member of the Acanthaceae family. Acanthaceae consists of about 220 genera and over 4000 species, and the genus Strobilanthes contains approximately 400 species (Deng et al. Citation2011).
The Strobilanthes species are essential herb sources. Strobilanthes cusia (Nees) Kuntze is the source plants of ‘Southern Ban-lan-gen’ in the Chinese Pharmacopeia (Pharmacopoeia Citation2015) with multiple pharmacological functions (Ko et al. Citation2006; Yu-Chi et al. Citation2020), particular for treating virus-induced diseases, such as SARS (Gu et al. Citation2015; Chia-Lin et al. Citation2019) and most recently, COVID-19. In contrast, Strobilanthes crispus has potent anticancer activities (Chin et al. Citation2017; Akowuah et al. Citation2020). Extracts of Strobilanthes species were also used to prevent animal and crop diseases (Manaf et al. Citation2016). For example, S. bantonensis has been used as a tea or animal feed additive to prevent fever (Lingfei Citation2016).
Strobilanthes bantonensis is mainly distributed in South China and Vietnam (Han et al. Citation2011). It has been used as a local herb in the Baise area (Shilin et al. Citation2011). It is also called ‘Purple Ban-lan-gen’ since the back of its leaf is purple, and has been traded as the herb ‘Southern Ban-lan-gen’ because their roots are very similar. However, there have been concerns regarding the equivalence in their pharmacological benefits. Consequently, distinguishing those two species is very important both in scientific research and the assurance of the effectiveness and safety of the herbal products.
The hypervariable sites from the chloroplast genome can be useful markers in discriminating closely related species (Lei et al. Citation2016). To distinguish S. cusia from other species in genus Strobilanthes, we had sequenced its chloroplast genome in a previous study (Chen et al. Citation2018). Here we report the complete chloroplast genome of S. bantonensis and hope this will help discriminate S. bantonensis from other plants of genus Strobilanthes and understand the evolutionary history of these plants.
2. Materials and methods
2.1. Sampling, DNA extraction, and sequencing
The fresh leaves of S. bantonensis were collected from Nianjing, Napo, Baise, Guangxi, China (Geospatial coordinate: E105.880213, N23.939937). The voucher samples were deposited in the Herbarium of the Guangxi botanical garden of medicinal plants (#451026101000LMS1). Total DNA was extracted using the plant genomic DNA kit and sequenced using the Hiseq2500 platform (Illumina, Inc., San Diego, CA).
2.2. Genome assembly and annotation
The chloroplast genome was assembled using NOVOPlasty (v. 2.7.2) (Dierckxsens et al. Citation2017) with a k-mer length of 39 bp and a conserved gene (rbcL gene) from Arabidopsis thaliana were used as the seed sequence. A total of 326,982 reads were used in the final plastid assembly and a circle genome was obtained. The average sequence coverage was 588.3. The clean reads were mapped to the assembled genome by using Bowtie2 (v.2.0.1) (Langmead 2012) for validating the correctness of the assembled chloroplast genome. The chloroplast genome was annotated using CPGAVAS2 (Shi et al. Citation2019) with the second option (2544-chloroplast genomes) of reference dataset. The results were further corrected manually using Apollo software (Lewis et al. Citation2002). The chloroplast genome sequence has been deposited in GenBank with the accession number (MT576695.1).
2.3. Repeats and SSR analyses
The GC content was calculated using the cusp program from EMBOSS (v. 6.3.1) (Rice et al. Citation2000). The simple sequence repeats (SSRs) were identified using MISA (https://webblast.ipk-gatersleben.de/misa/), including mono-, di-, tri-, tetra-, penta-, and hexanucleotides with the minimum numbers were 10, 6, 5, 5, 5, and 5, respectively (Beier et al. Citation2017). Additionally, REPuter (https://bibiserv.cebitec.uni-bielefeld.de/reputer/) was used to calculate palindrome repeats, forward repeats, reverse repeats, and complement repeats with the settings: Hamming Distance was three, and Minimal Repeat Size was 30 bp (Kurtz et al. Citation2001). Tandem repeats were detected with the Tandem Repeats Finder program (v. 4.07b, http://tandem.bu.edu/trf/trf.html) with default setting.
2.3. Genome comparison
We used MEGA (v. 6.0) (Tamura et al. Citation2013) to calculate the Codon usage of protein-coding sequences in S. bantonensis. RSCU (Relative Synonymous Codon Usage) value was calculated to analyze codon preference. The chloroplast genome sequences of S. cusia, were downloaded from GenBank (NC_037485.1). The two sequences were aligned by using MAFFT (v. 7.450) (Rozewicki et al. Citation2019), and the command was ‘mafft –thread 8 –threadtb 5 –threadit 0 –reorder –auto input > output.’ We conducted a sliding window analysis by using DnaSP (DNA Sequences Polymorphism, v. 6.0) to calculate the nucleotide polymorphism (Pi) among the species (window length: 600 bp, step size: 200 bp). Lastly, IRscope (https://irscope.shinyapps.io/irapp/) was used for visualizing the IR boundaries (Amiryousefi et al. Citation2018).
2.3. Phylogenetic analysis
The chloroplast genome sequences of 10 species belonging to the Acanthaceae family were downloaded from GenBank (Table S1). Two species, Nicotiana tabacum and Arabidopsis thaliana, were used as outgroups. A total of 68 orthologous genes among the 13 species were identified and extracted using Phylosuite (Zhang et al. Citation2020). They are ACCD, ATPA, ATPB, ATPE, ATPF, ATPH, ATPI, CCSA, CEMA, CLPP, MATK, NDHA, NDHB, NDHC, NDHD, NDHE, NDHF, NDHG, NDHH, NDHI, NDHJ, NDHK, PETA, PETB, PETD, PETG, PETL, PETN, PSAA, PSAB, PSAC, PSAJ, PSBA, PSBB, PSBC, PSBD, PSBE, PSBF, PSBH, PSBI, PSBJ, PSBK, PSBM, PSBT, PSBZ, RBCL, RPL14, RPL16, RPL20, RPL22, RPL23, RPL33, RPL36, RPOA, RPOB, RPOC1, RPS11, RPS14, RPS15, RPS16, RPS18, RPS2, RPS3, RPS4, RPS7, RPS8, YCF3, YCF4. The corresponding protein sequences were aligned using MAFFT (v 7.450) (Rozewicki et al. Citation2019) implemented in Phylosuite. These aligned sequences were used to construct the phylogenetic trees by using the maximum-likelihood (ML) method implemented in RaxML (v8.2.4) (Stamatakis Citation2014). The parameters were ‘raxmlHPC-PTHREADS-SSE3 -f a -N 1000 -m PROTGAMMALGX/GTRGAMMA -x 551314260 -p 551314260.’ The bootstrap analysis was performed with 1000 replicates.
Table 1. Basic features of the 2 chloroplast genomes.
3. Results
3.1. Structure of S. bantonensis chloroplast genome
A total of 6 G of raw sequencing data were generated. A chloroplast genome sequence was assembled successfully by using NOVOPlasty software. The correctness of the assembly was validated by mapping the raw sequence reads to the assembled genome and obtained a uniform coverage. The chloroplast genome of S. bantonensis is a circular DNA molecule with a total length of 144,591 bp. It has a conservative quadripartite structure, including a large-single copy (LSC) region, a small-single copy (SSC) region, and a pair of inverted repeat region (IR) regions, with the length of 92,068 bp, 17,767 bp, and 17,378 bp, respectively (). These results are similar to those of S. cusia, which is 91,666 bp, 17,811 bp, and 17,328 bp, respectively. We also compared the GC content of the two species. The total GC content is 38.18% in S. bantonensis, and 38.20% in S. cusia. The GC content in IR regions is much higher than that in the LSC or SSC region, consistent with the results observed in most angiosperms (Asaf et al. Citation2017).
3.2. Gene annotation
The chloroplast genome of S. bantonensis comprises 129 genes, includes 84 protein-coding, 37 tRNA, and eight rRNA genes ( and ). Ten protein-coding genes (rps16,atpF, rpoC1, petB, petD, rpl16, rpl2, ndhA and ndhB (×2) contain one intron, and two genes (ycf3, clpP) contain two introns. Eight tRNA genes (trnK-UUU, trnS-CGA, trnL-UAA, trnC-ACA, trnI-GAU (×2), trnA-UGC (×2)) contain one intron (). The length of the protein-coding, tRNA and rRNA genes in S. bantonensis chloroplast genome are 71,352 bp, 2813 bp, and 9078 bp respectively, accounting for 49.35%, 6.28%, and 1.95% of the total chloroplast genome length. Remarkably, a total of 16 genes replicated in the IR regions, of which, only 5 are protein-coding genes: psbA, ndhB, rps7, rps12, and ycf1 (only a fragment). This observation was less than that of other higher plants, suggesting that the IR regions may have undergone abnormal contraction or expansion.
Figure 1. Graphic representation of features identified in the chloroplast genome of S. bantonensis by using CPGAVAS2. The map contains four rings. From the center going outward, the first circle shows the forward and reverse repeats connected with red and green arcs. The next circle shows the tandem repeats marked with short bars. The third circle shows the microsatellite sequences identified using MISA. The fourth circle is drawn using drawgenemap and shows the gene structure on the chloroplast genomes. The genes were colored based on their functional categories, which are shown at the left corner.
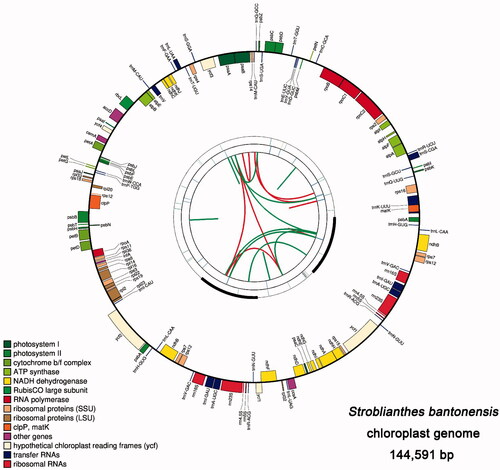
Table 2. Gene contents in the chloroplast genome of S. bantonensis.
Table 3. The lengths of introns and exons for the splitting genes.
3.3. Codon usage in protein-coding regions of the chloroplast genome
In terms of codon usage, a total of 23,784 codons were annotated in the chloroplast genome of S. bantonensis. The most common codon, AUU, codes for the amino acid Isoleucine (abbreviated I), was recorded 998 times (Table S2). Other common codes include AAA (936) and UUU (915), encoding lysine (abbreviated K), and Phenylalanine (abbreviated F), respectively. The relative synonymous codon use (RSCU) values are often used to assess the preference for codon usage in protein-coding. When RSCU value more than 1, it means that this codon is used preferentially. We observed that most amino acids have a codon preference except for the methionine (Met) and tryptophan (Trp) codons (Figure S1), which is similar to most higher plants (Sablok et al. Citation2011). In codons with high RSCU value, they usually end with A/T. For example, Leucine (Leu, RSCU = 1.87), Arginine (Arg, RSCU = 1.79), and Alanine (Ala, RSCU = 1.77) have a high preference for ‘UUA,’ ‘AGA,’ and ‘GCU,’ respectively. The high frequency of A/T usage in protein-coding regions may be the reason that the GC content is much lower than in tRNA and rRNA sequences.
3.4. Repeats analysis
We detected a total of 47 SSRs in the two analyzed species, and the results showed that most SSRs were mononucleotide repeats ( and Table S3). In the number of mononucleotide repeats, S. bantonensis is much higher than that of S. cusia. However, in the number of polynucleotide (di-, tri-) repeats, the results were reversed, which indicates that SSRs are highly polymorphism in both species. Detail analysis revealed that 23 SSR loci show polymorphism in these two species (Table S4), which could be potential cpSSR markers for species identification.
Figure 2. Comparison of the Repeats in the chloroplast genome of two Strobilanthes species. (a) Types and number of SSRs detected in the chloroplast genome sequences. (b) The number of tandem repeats and interspersed repeats.
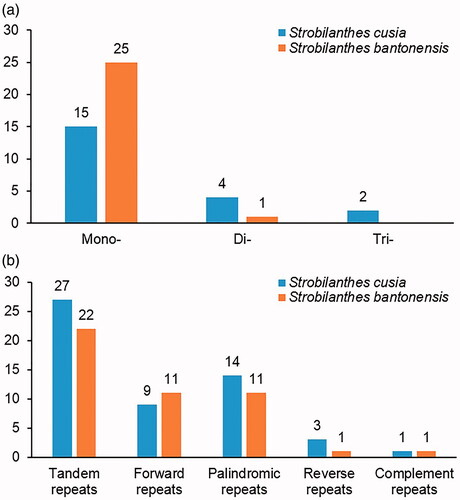
In the chloroplast genomes of the two Strobilanthes species, four types of interspersed repeats were detected. There are 9 forward repeats, 14 palindrome repeats, 3 reverse repeats, and only 1 complementary repeat in S. bantonensis. The numbers of interspersed repeats in S. cusia are 11, 11, 1, 1, respectively ( and Table S5). The numbers of these repeats exhibited significant interspecific differences. Besides, the number of tandem repeats is 27 in S. cusia and 22 in S. bantonensis, satisfying the conditions of length over 30 bp and similarity over 80% (Table S6). These sequences of tandem repeats and interspersed repeats are ubiquitous components of both prokaryotic and eukaryotic genomes (Heslop-Harrison Citation2000). It is also thought to be essential for promoting chloroplast genome rearrangements (McCann et al. Citation2018; Lee et al. Citation2019).
3.5. IR contraction event
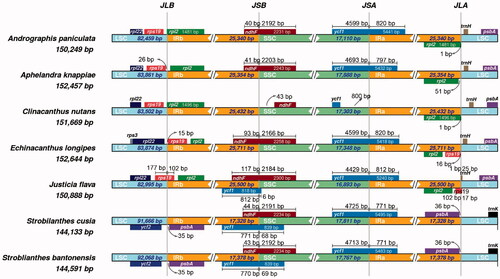
The contraction and expansion of IR regions are essential for the length diversity in chloroplast genomes (Goulding et al. Citation1996). In the chloroplast genomes of S. bantonensis and S. cusia, we observed a significant contraction of the IR regions (). The IR length of these two Strobilanthes species (∼17 kb) are both significantly shorter than those of the other four species from Acanthaceae family (∼25 kb), which suggests the occurrence of IR contraction events. The shrunk IR regions might cause the overall length of chloroplast genome in Strobilanthes being much shorter than those of other genera. Moreover, we observed a rare distribution of genes in the IR regions of S. bantonensis, which is very similar to that of S. cusia (Chen et al. Citation2018). In most angiosperms, the trnH and psbA genes are located in the LSC region, and the ycf2 gene is located in the IR regions with two copies. But in S. bantonensis and S. cusia, the gene, trnH, and psbA are found inside of the IR regions unexpectedly, while the ycf2 gene is found outside of the IR regions. So far, this particular distribution of genes in the IR regions has only been observed in these two Strobilanthes species and might be unique to this genus.
Figure 3. Comparison of the borders among LSC, SSC, and IR regions of seven species. The genes around the boundaries are shown above or below the main line. The JLB, JSB, JSA, and JLA represent junction sites of LSC/IRb, IRb/SSC, SSC/IRa, and IRa/LSC, respectively.
3.6. Divergence of chloroplast genome sequences
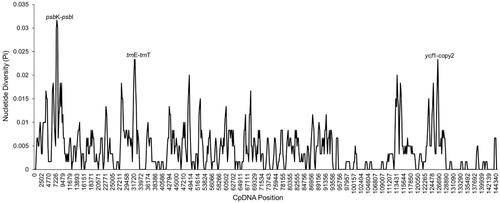
To evaluate the sequence divergence in the two Strobilanthes species, we used DnaSP software to quantify the levels of DNA polymorphism. There are three regions showed significant differences, which are psbK-psbI (0.03167), trnE-trnT (0.02333), and ycf1 (0.02333) (). The Pi values are listed in the parentheses, and they are all exceeding 0.02. These hypervariable regions could be used as potential barcodes to identify species.
Figure 4. Nucleotide diversity (Pi) for the two Strobilanthes species. There are two intergenic regions (psbK-psbI, 0. 03167; trnE-trnT, 0.02333 and one protein-coding region (ycf1, 0.02333) had Pi values greater than 0.02.
Furthermore, there are 605 variable single nucleotide sites (data not shown) and 116 indels (Table S7) in these two chloroplast genome sequences. Most indels involved only one base. We detected a deletion of 223 bp in the intergenic region rps16-trnQ-UUG in S. cusia. Besides, there are a total of 6 indels with a length of more than 30 bp.
We analyzed the variable sites in all protein-coding genes, and 32 of the 78 unique genes had no variable sites and were completely conserved (Table S8). The gene, ycf1, had the most variable sites (42), then followed by ycf2 (39), rpoC (17), and ccsA (14). Furthermore, the mutation rate for the three genes was more than 1%; they are ccsA (1.44%), ycf3 (1.19%), and rps15 (1.11%). Two protein-coding genes, matK (7, 0.7%) and rbcL (5, 0.35%), which are commonly used for DNA barcoding, are not able to distinguish the two species. In comparison, the gene ycf1 and ccsA look promising.
3.7. Phylogenetic analysis
To examine the phylogenetic position of S. bantonensis, we constructed the phylogenetic tree between S. bantonensis and 12 other species based on the datasets of chloroplast DNA sequences. As shown in , phylogenetic relationships based on the chloroplast genomes of 11 species of 6 genera of Acanthaceae resulted in the placement of S. bantonensis in the middle and upper part of the phylogenetic tree, with 100% bootstrap support.
Figure 5. Molecular phylogenetic analyses of chloroplast genomes in the tribe Heliantheae. The tree was constructed with the sequences of 68 proteins present in all 13 species using the maximum-likelihood method implemented in RAxML. Bootstrap supports were calculated from 1000 replicates. Nicotiana tabacum and Arabidopsis thaliana were set as outgroups. The number on the top of the branch indicates the bootstrap value. And the number at the bottom of the branch indicates the evolutionary distance. The genus to which each species belongs is shown to the right side of the tree.
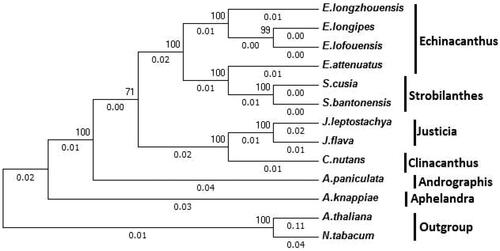
Aphelandra and Andrographis are at the base of the Phylogenetic trees in Acanthaceae. Then followed by Justicia and Clinacanthus. As the main clade, we concern about, S. bantonensis, and S. cusia, are get together as an unambiguous cluster in the phylogenetic tree and form a monophyletic group. Echinacanthus was the closest taxa to Strobilanthes.
However, due to the maternal inheritance of the plastid genome, these results are limited. Accurate phylogenetic relationships still require a comprehensive analysis of nuclear and organellar genes. The phylogenetic relationships of E. attenuates and Strobilanthes species require further study. Furthermore, more genome sequencing are needed in the future to determine the relationships among Strobilanthes and other species from the family Acanthaceae.
4. Discussion
In summary, we assembled and analyzed a complete chloroplast genome with data generated by next-generation DNA sequencing for Strobilanthes bantonensis. One of the most interesting observations is the peculiar structure of the IR regions.
Recently, five chloroplast genomes from species of genus Dicliptera (Acanthaceae) were sequenced and analyzed (Huang et al. Citation2020). This study is the first systematic analysis of the completed chloroplast genomes of a genus in Acanthaceae's large family. We had reported the first chloroplast genomes of genus Strobilanthes of the family Acanthaceae (Chen et al. Citation2018), and find its Inverted Repeat Region (∼17 kb) is much shorter than that of most other angiosperms (∼20–28 kb) (Chumley et al. Citation2006). However, the IR region of the genus Dicliptera is ∼25kb, which means the IR contraction is not the characteristics of family Acanthaceae. Here we find that the IR region of S. bantonensis is ∼17 kb, similar to that of the S. cusia, suggesting IR contraction may be a characteristic feature of the genus Strobilanthes.
The phylogenetic tree we have constructed is basically consistent with previous research (Ding et al. Citation2016; Li et al. Citation2019; Yaradua et al. Citation2019). Surprisingly, although Echinacanthus was a sister group to Strobilanthes, E. attenuates appears to be closer to Strobilanthes with high bootstrap values than other Echinacanthus species. This is consistent with the previous study (Gao et al. Citation2018), and this implies that Echinacanthus may not be a monophyletic group. It seems that E. attenuates should be classified as a species of Strobilanthes, and Echinacanthus and Strobilanthes may be more closely related than we previously thought.
The over 400 Strobilanthes species represent a rich source for bioprospecting. Correct identification of these species is a critical step. However, the Strobilanthes species are very similar in morphological characters and highly homoplasious, thus causing difficulty in further classification and differentiation. Different methods have been used to discriminate plants from the Strobilanthes genus, including morphological investigation, chemical analysis, and gene identification (Ni et al. Citation2012). DNA barcoding is one of the most promising methods. However, the single locus barcode does not have as much genetic information as the chloroplast genome for species discrimination. We are currently collecting a large number of Strobilanthes samples to test molecular markers predicted from this study.
Authors’ contributions
W.W. and C.L. conceived and designed this study. Y.L. and M.S.L. collected the samples. J.J. extracted DNA for next-generation sequencing. M.J. assembled the complete chloroplast genome. W.W. and J.J.L. annotated and analyzed the chloroplast genome and wrote the manuscript. All authors have read the work and agreed with its contents.
Data availability statement
The scientific name of the organism is Strobilanthes bantonensis Lindau. The specimen was collected in Nianjing, Napo, Baise, Guangxi, China (Geospatial coordinate: E105.880213, N23.939937). The specimen and its DNA are stored in the Herbarium of the Guangxi botanical garden of medicinal plants (#451026101000LMS1). The data that support the findings of this study are openly available in NCBI at https://www.ncbi.nlm.nih.gov/nuccore/MT576695.
Additional information
Funding
References
- Akowuah GA, Chin JH, Yeong SW, Quah SY, Ahmad M. 2020. In-vitro CYP3A4 , CYP2E1 and UGT Activity in Human Liver Microsomes by Strobilanthes crispus Leaf Extracts. NPJ. 10(2):104–112.
- Amiryousefi A, Hyvönen J, Poczai P. 2018. IRscope: an online program to visualize the junction sites of chloroplast genomes. Bioinformatics. 34(17):3030–3031.
- Asaf S, Khan AL, Khan MA, Waqas M, Kang SM, Yun BW, Lee IJ. 2017. Chloroplast genomes of Arabidopsis halleri ssp. gemmifera and Arabidopsis lyrata ssp. petraea: structures and comparative analysis. Sci Rep. 7(1):7556.
- Beier S, Thiel T, Münch T, Scholz U, Mascher M. 2017. MISA-web: a web server for microsatellite prediction. Bioinformatics. 33(16):2583–2585.
- Chen H, Shao J, Zhang H, Jiang M, Huang L, Zhang Z, Yang D, He M, Ronaghi M, Luo X. 2018. Sequencing and analysis of Strobilanthes cusia (Nees) Kuntze chloroplast genome revealed the rare simultaneous contraction and expansion of the inverted repeat region in angiosperm. Front Plant Sci. 9:324.
- Chia-Lin L, Chien-Ming W, Hao-Chun H, Hung-Rong Y, Ying-Chyi S, Sheng-Jie Y, Yang-Chang W. 2019. Indole alkaloids indigodoles A-C from aerial parts of Strobilanthes cusia in the traditional Chinese medicine Qing Dai have anti-IL-17 properties. Phytochemistry. 162: 39–46.
- Chin JE, Ikhtifar MRM, Khoo CH, Ho KL, Cheah YK. 2017. Analysis of chemical constituents, antimicrobial and anticancer activities of dichloromethane extracts of Sordariomycetes sp. endophytic fungi isolated from Strobilanthes crispus. World J Microbiol Biotechnol. 33(1):5.
- Chumley TW, Palmer JD, Mower JP, Fourcade HM, Calie PJ, Boore JL, Jansen RK. 2006. The complete chloroplast genome sequence of Pelargonium x hortorum: organization and evolution of the largest and most highly rearranged chloroplast genome of land plants. Mol Biol Evol. 23(11):2175–2190.
- Deng Y, J.-Q H, Daniel TF, Wood J, Wood JRI. 2011. Acanthaceae. In: Flora of China. Vol. 19. Beijing: Science Press.
- Dierckxsens N, Mardulyn P, Smits G. 2017. NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 45(4):e18.
- Ding P, Shao Y, Li Q, Gao J, Zhang R, Lai X, Wang D, Zhang H. 2016. The complete chloroplast genome sequence of the medicinal plant Andrographis paniculata. Mitochondrial DNA A DNA Mapp Seq Anal. 27(4):2347–2348.
- Gao C, Deng Y, Wang J. 2018. The complete chloroplast genomes of Echinacanthus species (Acanthaceae): phylogenetic relationships, adaptive evolution, and screening of molecular markers. Front Plant Sci. 9:1989.
- Goulding SE, Olmstead RG, Morden CW, Wolfe KH. 1996. Ebb and flow of the chloroplast inverted repeat. Mol Gen Genet. 252(1-2):195–206.
- Gu W, Wang W, Li X-N, Zhang Y, Wang L-P, Yuan C-M, Huang L-J, Hao X-J. 2015. A novel isocoumarin with anti-influenza virus activity from Strobilanthes cusia. Fitoterapia. 107:60–62.
- Han X, Mingxian L, Yide L. 2011. Strobilanthes bantonensis Lindau, a newly recorded species of Acanthaceae from Hainan, China. J Tropical Organ. 2(4):302–304.
- Heslop-Harrison JS. 2000. Comparative genome organization in plants: from sequence and markers to chromatin and chromosomes. Plant Cell. 12(5):617–636.
- Huang S, Ge X, Cano A, Salazar BGM, Deng Y. 2020. Comparative analysis of chloroplast genomes for five Dicliptera species (Acanthaceae): molecular structure, phylogenetic relationships, and adaptive evolution. PeerJ. 8:e8450.
- Ko HC, Wei BL, Chiou WF. 2006. The effect of medicinal plants used in Chinese folk medicine on RANTES secretion by virus-infected human epithelial cells. J Ethnopharmacol. 107(2):205–210.
- Kurtz S, Choudhuri JV, Ohlebusch E, Schleiermacher C, Stoye J, Giegerich R. 2001. REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 29(22):4633–4642.
- Lee HO, Joh HJ, Kim K, Lee SC, Kim NH, Park JY, Yang TJ. 2019. Dynamic chloroplast genome rearrangement and DNA barcoding for three Apiaceae species known as the medicinal herb “Bang-Poong.” Int J Mol Sci. 20(9): 2196.
- Lei W, Ni D, Wang Y, Shao J, Wang X, Yang D, Wang J, Chen H, Liu C. 2016. Intraspecific and heteroplasmic variations, gene losses and inversions in the chloroplast genome of Astragalus membranaceus. Sci Rep. 6:21669.
- Lewis SE, Searle SMJ, Harris N, Gibson M, Lyer V, Richter J, Wiel C, Bayraktaroglu L, Birney E, Crosby MA. 2002. Apollo: a sequence annotation editor. Genome Biol. 3(12):research0082–0014.
- Li M-N, Pei H-Q, Wang J-H, Zhao K-K, Zhu Z-X, Wang H-F. 2019. Complete plastome sequence of Clinacanthus nutans (Acanthaceae): a medicinal species in Southern China. Mitochondrial DNA B. 4(1):118–119.
- Lingfei G. 2016. “Hot" and "Cold" of purple Economy (Master). Nanning (Guangxi): Guangxi University for Nationalities.
- Manaf SR, Manaf SR, Daud HM, Daud HM, Alimon AR, Mustapha NM, Hamid NH. 2016. The effects of Vitex trifolia, Strobilanthes crispus and Aloe vera herbal-mixed dietary supplementation on growth performance and disease resistance in red hybrid Tilapia (Oreochromis sp.). J Aquacult Res Dev. 7(4):425.
- Mccann J, Jang T-S, Macas J, Schneeweiss GM, Matzke NJ, Novák P, Stuessy TF, Villaseñor JL, Weiss-Schneeweiss H. 2018. Dating the species network: allopolyploidy and repetitive DNA evolution in American Daisies (Melampodium sect. Melampodium, Asteraceae). Syst Biol. 67(6):1010–1024.
- Ni Y, Song R, Kokot S. 2012. Discrimination of Radix Isatidis and Rhizoma et Radix Baphicacanthis Cusia samples by near infrared spectroscopy with the aid of chemometrics. Spectrochim Acta A Mol Biomol Spectrosc. 96:252–258.
- Pharmacopoeia. 2015. Pharmacopoeia of the people's Republic of China. Vol. 1. Beijing: China Medical Science Press.
- Rice P, Longden I, Bleasby A. 2000. EMBOSS: the European molecular biology open software suite. Trends Genet. 16(6):276–277. TIG,
- Rozewicki J, Li S, Amada KM, Standley DM, Katoh K. 2019. MAFFT-DASH: integrated protein sequence and structural alignment. Nucleic Acids Res. 47(W1):W5–W10.
- Sablok G, Nayak KC, Vazquez F, Tatarinova TV. 2011. Synonymous codon usage, GC(3), and evolutionary patterns across plastomes of three pooid model species: emerging grass genome models for monocots. Mol Biotechnol. 49(2):116–128.
- Shi L, Chen H, Jiang M, Wang L, Wu X, Huang L, Liu C. 2019. CPGAVAS2, an integrated plastome sequence annotator and analyzer. Nucleic Acids Res. 47(W1):W65–W73.
- Shilin S, Tingting Z, Bo M, Ke H, Xiaobiao Z. 2011. Study on the ethnobotany in the minority area of Baise Region. Anhui Agric Sci Bull. 17(23):123.
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30(9):1312–1313.
- Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. 2013. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 30(12):2725–2729.
- Yaradua SS, Alzahrani DA, Albokhary EJ, Abba A, Bello A. 2019. Complete chloroplast genome sequence of Justicia flava: genome comparative analysis and phylogenetic relationships among Acanthaceae. Biomed Res Int. 2019:1–17.
- Yu-Chi T, Chia-Lin L, Hung-Rong Y, Young-Sheng C, Yu-Ping L, Su-Hua H, Cheng-Wen L. 2020. Antiviral action of Tryptanthrin isolated from Strobilanthes cusia leaf against human coronavirus NL63. Biomolecules. 10(3):366.
- Zhang D, Gao F, Jakovlić I, Zou H, Zhang J, Li WX, Wang GT. 2020. PhyloSuite: an integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol Ecol Resour. 20(1):348–355.