
Abstract
Lactuca raddeana is a biennial plant of the Lactuca genus belonging to the Asteraceae family. The classification of Lactuca is controversial, and no consistent conclusions have been reached based on the analysis of morphological characters and different molecular markers. Here, we sequenced and assembled the complete chloroplast genome of L. raddeana. This genome has a total length of 152,339 bp, a conservative quartile structure that is composed of a large single-copy (LSC) region of 83,976 bp, a small-copy (SSC) region of 18,521 bp, and a pair of inverted repeats (IRs) of 24,921 bp. The genome contains 112 unique genes, including 79 protein-coding, four rRNA, and 29 tRNA genes. Repeat analysis obtained 17 microsatellite, 16 tandem, and 17 interspersed repeats. Comparison of sequence divergence between L. raddeana and L. sativa found the intergenic spacer trnC-petN exhibited the highest degree of variation. Three phylogenetic trees based on the 72 shared protein, matK gene, and rbcL gene sequences showed that L. raddeana and L. sativa are clustered together. The acquisition and comparative analysis of the chloroplast genome provide a valuable resource for the taxonomic and phylogenetic studies of Lactuca.
Introduction
Lactuca raddeana belongs to the genus Lactuca, family Asteraceae, distributed in China, Russia, North Korea, Japan, and South Korea. It grows in forests, forest margins, thickets, moist areas on mountain slopes, mountain valleys, fields, and trailside (iPlant Citation2020a). The Lactuca genus includes at least 50 species, distributed worldwide, but mainly in temperate Eurasia (iPlant Citation2020b). For L. raddeana, it is found that its chemical component lactuside B improved ischemic brain injury by reducing aquaporin-4 and caspase-3 mRNA expression in the hippocampus and striatum three and regulating the expression of BCL-2 and BAX mRNA (Li et al. Citation2011). However, the effects of other species from this genus remained to be discovered. Clarification of the taxonomic classification of L. raddeana is critical for the future development of new drugs based on L. raddeana.
The classification of Lactuca is controversial. L. raddeana has several alias names, including Pterocypsela elata (Hemsl.) Shih, Pterocypsela raddeana (Maxim.) Shih, Lactuca vaniotii H. Lév., Lactuca alliariifolia H. Lév. et Vaniot, Lactuca elata Hemsl., Lactuca raddeana Maxim. var. elata (Hemsl.) Kitam. and Prenanthes hieraciifolia H. Léveillé (iPlant, Citation2020a), reflecting the difficulty in determining its taxonomy classification using morphological characters. More recently, the nuclear rDNA ITS sequences and five chloroplast DNA markers had been used to delineate the phylogeny of Lactuca species. Unfortunately, inconsistent results were obtained using these markers (Wang et al. Citation2013). The phylogeny of Lactuca was analyzed based on 31 chloroplast genomes (LSC + SSC + IR), nuclear rDNA, and ITS sequences, indicating that there are at least four main groups (Wei Citation2016). However, these species cover just 36% of the Lactuca, and there are still some differences among the three trees. Thus, more phylogenetic analysis based on chloroplast genomes is needed to study the phylogeny of Lactuca furtherly.
The chloroplast is an essential organelle in plant cells and capable of photosynthesis in the presence of light. The complete chloroplast genome provides a rich resource for accurate identification of plant species and determining their phylogenetic relationships (Daniell et al. Citation2016). Chloroplast genome encodes many key proteins playing essential roles in photosynthesis and other metabolic processes (Keeling Citation2004). The structure of the chloroplast genome is highly conserved and usually consists of four parts with a large single-copy (LSC) region, a small single-copy (SSC) region, and two inverted repeats (IRs) (Palmer Citation1991). The chloroplast genome sequence can be divided into the protein-coding region and the non-coding region. The coding region has a slow evolution rate and is suitable for phylogenetic analysis of higher taxonomic levels such as order and family (Li et al. Citation2012). The non-coding region evolves faster and has more variation information, which is more suitable for phylogenetic analysis of lower taxonomic levels such as genus and species (Shaw et al. Citation2007).
Previously, the partial chloroplast genome sequence of Lactuca raddeana has been reported, which has 127,660 bp containing an LSC, an SSC, and one IR region (Wei Citation2016). However, this genome sequence was not publicly available. To address this problem, we sequenced, assembled, and annotated the complete chloroplast genome sequence of L. raddeana and compared it with the chloroplast genomes of L. sativa.
Material and methods
Plant material, DNA extraction, and sequencing
We collected fresh leaf samples from the Central China Medicinal Botanical Garden, EnShi, China (Geospatial coordinates: N30.178176, E107.745725) and identified them as L. raddeana by Professor Jinwen You. We kept the sample in the −80° refrigerator until use. We extracted the genomic DNA with plant genomic DNA kit (Tiangen Biotech, China) and constructed the DNA library with 1 ug DNA using the library preparation kit (New England BioLabs. America). We sequenced the library using the Hiseq 2500 platform (Illumina, America). A total of 26,321,582 paired-end reads (2 × 150 bp) were obtained. Clean data were obtained by removing low-quality sequences: the percentage of bases with a quality value of Q < 19 is more than 50%, and the percentage of ‘N’ is more than 5%.
Genome assembly and annotation
We assembled the clean sequence data into a chloroplast genome by NOVOPlasty (v. 2.7.2) (Dierckxsens et al. Citation2017) with the k-mer length of 39 bp and the sequence of rbcL from Arabidopsis thaliana as the seed. We manually examined the assembly by mapping all raw reads to the assembled genome sequence using Bowtie2 (v.2.0.1) (Langmead et al. Citation2009) under the default settings. Complete coverage and an even sequencing depth across the assembly would support the correctness of the assembly. The annotation of the chloroplast genome was initially performed using CpGAVAS2 (Shi et al. Citation2019), and then the annotations with problems were edited using Apollo (Misra and Harris Citation2006). The genome sequence and annotations have been deposited in GenBank with accession number MN402448.
General characteristics and repeat analysis
The codon usage and repeat analysis were analyzed using CpGAVAS2. Notably, we identified the microsatellite repeats with MISA software (Beier et al. Citation2017). And the cutoff for the numbers of units for mono-, di-, tri-, tetra-, Penta-, and hexanucleotides were 10, 6, 5, 5, 5, and 5, respectively. We identified the tandem repeats by using TRF software (Benson Citation1999) with the size of repeat unit > = 7. Then, we identified the interspersed repeats with VMATCH software (Kurtz et al. Citation2001). Lastly, we calculated both GC contents and codon usage using the program Cusp from EMBOSS (v6.3.1) (Rice et al. Citation2000).
Comparative analysis of the chloroplast genomes
The comparative analysis between L. raddeana and the L. sativa was conducted using the mVISTA program (Frazer et al. Citation2004) in a Shuffle-LAGAN mode with default parameters. The complete chloroplast genome of L. sativa (Accession number: NC_007578) was obtained from Genbank. And the annotated L. raddeana genome was used as the reference. The IR boundary was analyzed and visualized by using the IRscope tool (Amiryousefi et al. Citation2018).
Phylogenetic analysis
We constructed the phylogenetic tree between the L. raddeana and other fourteen closely related species obtained from Genbank (Table S4), L. sativa, Sonchus acaulis, Sonchus boulosii, Sonchus canariensis, Sonchus webbii, Taraxacum amplum, Taraxacum brevicorniculatum, Taraxacum kok-saghyz, Taraxacum mongolicum, Taraxacum obtusifrons, Taraxacum officinale, Taraxacum platycarpum, Youngia japonica, and Youngia denticulata. Helianthus annuus and Ageratina adenophora were set as outgroups. A total of 72 shared proteins present among the sixteen chloroplast genomes were detected and extracted (ATPA, ATPB, ATPE, ATPF, ATPH, ATPI, CCSA, CEMA, CLPP, MATK, NDHA, NDHB, NDHC, NDHD, NDHE, NDHF, NDHG, NDHH, NDHI, NDHJ, NDHK, PETA, PETB, PETD, PETG, PETL, PETN, PSAA, PSAB, PSAC, PSAI, PSAJ, PSBA, PSBB, PSBC, PSBD, PSBE, PSBF, PSBH, PSBJ, PSBK, PSBM, PSBN, PSBT, RBCL, RPL14, RPL16, RPL2, RPL20, RPL22, RPL23, RPL32, RPL33, RPL36, RPOA, RPOB, RPOC1, RPOC2, RPS11, RPS12, RPS14, RPS15, RPS16, RPS18, RPS19, RPS2, RPS3, RPS4, RPS7, RPS8, YCF3, YCF4). These protein sequences were subjected to multiple sequence alignment using CLUSTALW2 (v2.0.12) (Thompson et al. Citation2002) with the option ‘-gapopen = 10 -gapext = 2 -output = phylip’. The phylogenetic tree was then constructed using the Maximum Likelihood method implemented in RaxML (v8.2.4) (Stamatakis Citation2014) with the option ‘raxmlHPC-PTHREADS-SSE3 -f a -N 1000 -m PROTGAMMACPREV -x 551314260 -p 551314260 -o A_thaliana, N_tabacum -T 20’.
Results
General features of the chloroplast genome
The chloroplast genome of L. raddeana is 152,339 bp in size with an LSC of 83,976 bp, an SSC of 18,521 bp, and two IRs of 24,921 bp each (). The chloroplast genome encodes 130 genes, of which 112 are unique genes, including 79 protein-coding genes, four ribosome RNA genes, and 29 transfer RNA genes (). Among them, there are 7 genes (rps16, rpoC1, atpF, petB, rpl2, ndhB, and ndhA) contains 1 intron, 2 genes (ycf3 and clpP) contain 2 introns. 6 tRNAs (trnK-UUU, trnS-CGA, trnL-UAA, trnC-ACA, trnE-UUC, and trnA-UGC) contain 1 intron (Table S1). The length of the protein-coding regions, the rRNA genes, and the tRNA genes in the chloroplast genome of the L. raddeana is 78,288 bp, 9,536 bp, and 2,727 bp, accounting for 51.39%, 6.26%, and 1.79% of the genome length, respectively. The chloroplast genome's non-coding region mainly includes introns and intergenic spacers, and its length accounts for 40.56% of the genome length.
Figure 1. The schematic representation of the chloroplast genome of L. raddeana was created by using CPGAVAS2. The map contains four rings. From the center going outward, the first circle shows the scattered forward and reverse repeats connected with red and green arcs, respectively. The next circle shows the tandem repeats marked with short bars. The third circle shows the microsatellite sequences identified. The fourth circle shows the gene structure of the chloroplast genome. The genes were colored based on their functional categories, which are shown in the left corner.
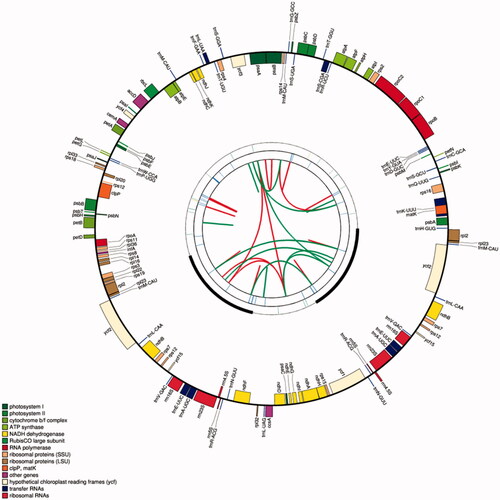
Table 1. Gene composition in the chloroplast genome of L. raddeana.
The whole genome's GC content is 37.69%, of which the protein-coding regions, the rRNA genes, and the tRNA genes are 38.02%, 54.55%, and 53.12%, respectively. The GC contents for the first, second, and third positions of the codons are 45.58%, 38.26%, and 30.22%, respectively, showing a higher A/T bias within the protein-coding regions at the third codon position. In terms of codon usage, 26,096 codons encoding 79 protein-coding genes were identified in the complete genome (Table S2). The most common codon, AUU, codes for the amino acid Isoleucine (an abbreviation I) and was found 1,056 times.
Table 2. Microsatellite repeats in the chloroplast genome of L. raddeana.
Repeat analysis
The types and numbers of repeated sequences might provide important information regarding genome evolution. There are three types of repeats. The simple sequence repeats (SSRs), also referred to as microsatellite repeats, consist of multiple copies of small repeat units (size <= 6 bp) concatenated together. They are abundant in the angiosperm genomes and often used as molecular markers because they mutate rapidly at the interspecies and intraspecies levels (Lu et al. Citation2005; Tanaka et al. Citation2017). In the chloroplast genome of L. raddeana, the SSRs include 15 A/T and 2 AT/AT (). These numbers are rather small compared to those found in other chloroplast genomes (data not shown). The SSRs were found to distribute across the intergenic spacers (IGS), intron regions, and exon regions. And numbers of SSRs falling into these regions are 9, 3, and 3, respectively.
The second type of repeats is the tandem repeat consisting of multiple copies of repeat units (size > 6 bp) concatenated together. A total of 26 tandem repeats were found in the chloroplast genome of L. raddeana, of which the most extended repeat unit is 32 bp (). And the number of repeat units is mostly 2. The average matching rate of repeat unit sequences is 98%. Seventeen of the tandem repeats possessed the highest possible matching rate of 100%. Besides, four tandem repeats are present in the IR regions and located in the IGS (trnN-GUU-trnR-ACG and trnN-GUU-trnR-ACG) and exonic (rrn4.5S and ycf2) regions.
Table 3. Tandem repeats in the chloroplast genome of L. raddeana.
The third type of repeats is the interspersed repeat. It differs from the tandem repeat in the organization. Remarkably, the repeat units are distributed in a scattered and nonadjacent manner within the genome. We identified two tandem repeats in the chloroplast genome, 9 palindromic repeats, and 16 direct repeats (Table S3). The repeats sequences of three types in the chloroplast genome of L. raddeana will be invaluable in developing molecular markers.
Comparative genomic analysis
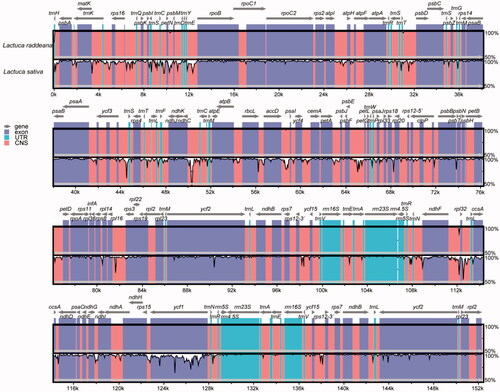
To investigate the similarities and divergence between L. raddeana and other Lactuca plants' chloroplast genomes, we compared the sequence divergence between the chloroplast genomes of L. raddeana and the L. sativa (). In general, the two genomes are similar, in which the protein-coding regions are more conserved than the non-coding regions. Specifically, the ycf1 genes from the two species are highly variable, consistent with the previous finding that the ycf1 gene exhibited a rapid evolution rate in many species. For the non-coding regions, seven regions have a higher degree of divergence, with the percentage of similarity being less than 75%. These include IGS(trnC-petN), IGS(trnE-rpoB), IGS(ycf3-trnS), IGS(ndhC-trnM), IGS(psbF-petG), IGS(rpl16-rps3), IGS(rpl32-trnL).
Figure 2. Comparison of the chloroplast genomes of L. raddeana and L. sativa by using mVASTA. The gray line and arrow indicated the orientation of genes. The coloring of the different conserved regions corresponds to the annotation of the region. The genes were represented as gray arrows on the top of the alignment. Other regions are labeled with different colors. The pink regions are ‘Conserved Non-Coding Sequences’ (CNS), the dark blue regions are exons, and the light-blue regions are ‘Untranslated regions’ (UTR). The Y-scale represents the percent identity ranging from 50% to 100%.
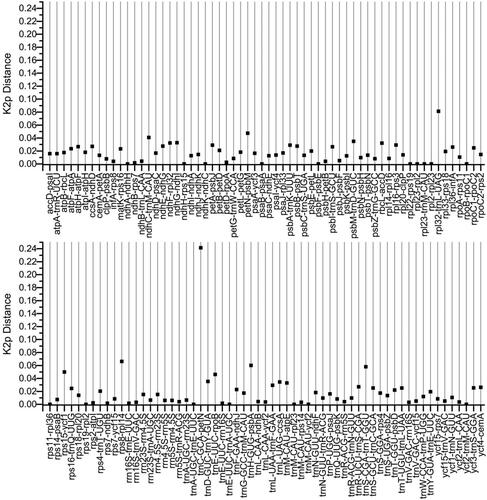
The distances of intergenic spacers between the L. raddeana and L. sativa were conducted using the Kimura 2-parameter (K2p) model (). The K2p distances range from 0.00-0.242, with an average of 0.018. The seventeen IGS regions: ndhA-ndhH, ndhH-rps15, ndhK-ndhC, psaB-psaA, psbF-psbE, rpl22-rps19, rpl23-rpl2, rpl23-trnM, rpl2-rpl23, rpoB-rpoC1, rps11-rpl36, rps19-rpl2, trnA-trnE, trnE-trnA, trnM-rpl23, trnM -ycf2, ycf2-trnM, exhibiting 100% similarity with the K2p values of 0. These IGS regions are conserved and have a length range from 1 bp to165bp. Besides, the IGS(trnC-petN) has the largest K2p values of 0.242, which indicated a faster evolution rate than other regions. The length of IGS(trnC-petN) varies greatly, 534 bp for L. raddeana and 831 bp for L. sativa. In summary, from the highly variable region, it is expected to develop new DNA barcodes to discriminate different species of Lactuca.
Figure 3. K2p distances between the IGS regions of L. raddeana and L. sativa.
Structure analysis of the IR boundaries
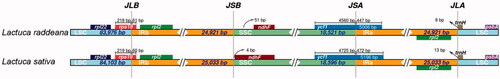
The contraction and expansion of IR boundaries are often related to species evolution and directly affect the chloroplast genome's size. Previous studies have shown that the length and boundary of the IR region are variable in different species. Here the IR boundaries of L. raddeana and L. sativa chloroplast genomes were analyzed, showing that they have similar boundary structures (). The LSC-IRb and SSC-IRa boundaries are located in the protein-coding regions, the rps19, and the ycf1 genes. In contrast, the IRb-SSC and IRa-LSC boundaries are located in the IGS, the IGS(trnN -ndhF), and IGS(rpl2-trnH), respectively. It should be pointed out that not all genes located in the IR regions are duplicated. For example, there is only one ycf1 gene since part of the ycf1 gene is located in the IRa region, and the other part is in the SSC region. Compared with other angiosperms, the Lactuca genus' IR boundaries were relatively conservative and had no significant contraction and expansion (Chen et al. Citation2018).
Figure 4. LSC, SSC, and IR regions' boundaries in the chloroplast genomes of L. raddeana and L. sativa. The genes on the positive and negative strands are presented above and below the tracks, respectively. The JLB, JSB, JSA, and JLA represent junction sites of IRb and LSC, IRb and SSC, SSC and IRa, and IRa and LSC, respectively.
Phylogenetic analysis
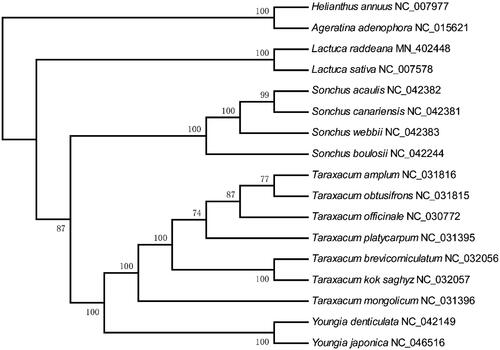
The chloroplast genome provides a valuable resource for the phylogenetic and taxonomic studies of the angiosperm (Guo et al. Citation2017). We obtained fifteen complete chloroplast genome sequences to explore the phylogenetic relationships among species from Cichorieae. The sequences of 72 shared proteins were used to construct the phylogenetic tree. In general, the species belong to the same genus were clustered together (). The genus Taraxacum, Sonchus, and Youngia were clustered together, and the Lactuca species formed a single phylogenetic cluster. Some taxonomists classified the L. raddeana as a species of Pterocypsela Shih's genus, and the classification of the genus Pterocypsela Shih and Lactuca themselves is highly controversial (Wang et al. Citation2013). In this study, L. raddeana and L. sativa were clustered together with 100 bootstrap value, which supports L. raddeana belongs to the Lactuca genus.
Figure 5. The phylogenetic tree of species from Cichorieae constructed using the maximum likelihood (ML) method using the 72 shared proteins present among the seventeen chloroplast genomes. The number above each node represents are the bootstrap value.
Discussion
In this study, we sequenced and analyzed the complete chloroplast genome of L. raddeana. Specifically, we extracted and sequenced the total DNA from the leaves of L. raddeana, assembled and annotated a complete chloroplast genome. Then we analyzed the general features of the chloroplast genome and the three kinds of repeat sequences, compared the genomic divergence and IR boundaries between the L. raddeana and L. sativa, and conducted phylogenetic analyses of L. raddeana and several closely related species from the Cichorieae. The present study provides a high-quality reference chloroplast genome sequence for the L. raddeana, which can be used for phylogenetic analyses of Lactuca species.
The Lactuca belong to the tribe Cichorieae, family Asteraceae. Due to the ambiguous definition of the genus, the number of species given in the literature varies greatly. The traits between Lactuca and its related genus are complex and have a serious crossover. There are two main types in the classification of Lactuca according to morphological characteristics. The first type, the Cicerbita Wallr., Mycelis Cass., Mulgedium Cass., and Pterocypsela, is combined as the Lactuca genus section (Lebeda et al. Citation1999; Doležalová et al. Citation2002). The second type, the Cicerbita Wallr., Mycelis Cass., Mulgedium Cass., and Pterocypsela, is considered an independent genus (Chu Citation1988).
The Lactuca raddeana of this study is also named Pterocypsela raddeana in the same literature (Wang et al. Citation2013). This study constructed a phylogenetic tree based on the shared protein sequence, showing that L. raddeana and L. sativa are clustered together with 100 bootstrap values. Previous studies based on different molecular sequences have shown different phylogenetic results. The phylogenetic analysis based on the entire chloroplast genome sequence without one of the IR regions showed that the Pterocypsela group, including L. raddeana, was clustered together with those of the crop group L. sativa (Wei Citation2016). The phylogenetic analysis based on petD gene, IGS(psbA-trnH), IGS(trnL-UAA-trnF), IGS(rpl32-trnL-UAG), and IGS(trnQ-UUG-rps16) showed that the genus Pterocypsela including L. raddeana and Lactuca formed two separate branches (Wang et al. Citation2013). The phylogenetic tree based on ITS region indicated that the Pterocypsela (including L. raddeana) and some of the Lactuca species (including L. sativa) were on one branch (Wang et al. Citation2013).
What’s more, the L. raddeana are not clustered together with L. sativa according to the tree constructed using the ndhF and trnL genes, but on the same branch with other Lactuca species (Wei et al. Citation2017). In summary, the study based on part and complete chloroplast genome supported the notion that the Lactuca raddeana (Pterocypsela raddeana) belongs to the Lactuca genus. However, the current study cannot resolve the classification of all the Lactuca species due to limited numbers of chloroplast genomes available. More chloroplast genome sequences are needed to determine the phylogeny of these species ultimately.
Authors’ contributions
CL conceived the study; MJ collected samples of L. raddeana, extracted DNA for next-generation sequencing, assembled and validated the genome; YNL performed data analysis, and drafted the manuscript. BW, HMC, and CL critically reviewed the manuscript. All authors have read and agreed on the contents of the manuscript.
Disclosure statement
The authors declare that they have no competing interests.
Data availability statement
The data that support the findings of this study are openly available in NCBI (National Center for Biotechnology Information). The accession number of the annotated chloroplast genome is MN402448 (https://www.ncbi.nlm.nih.gov/nuccore/MN402448). The accession number of the raw sequence data is PRJNA688122. The sample has been deposited in the Herbarium of the Institute of Medicinal Plant Development in Beijing, China, with the accession number: implad201808023.
Additional information
Funding
References
- Amiryousefi A, Hyvönen J, Poczai P. 2018. IRscope: an online program to visualize the junction sites of chloroplast genomes. Bioinformatics. 34(17):3030–3031.
- Beier S, Thiel T, Munch T, Scholz U, Mascher M. 2017. MISA-web: a web server for microsatellite prediction. Bioinformatics. 33(16):2583–2585.
- Benson G. 1999. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27(2):573–580.
- Chen H, Shao J, Zhang H, Jiang M, Huang L, Zhang Z, Yang D, He M, Ronaghi M, Luo X, et al. 2018. Sequencing and analysis of Strobilanthes cusia (Nees) Kuntze chloroplast genome revealed the rare simultaneous contraction and expansion of the inverted repeat region in angiosperm. Front Plant Sci. 9:324., (2018).
- Chu S. 1988. Revision of Lactuca L. and two new genera of Tribe lactuceae (Compositae) on the mainland of Asia. J Syst Evol. 26(5):382–393.
- Daniell H, Lin CS, Yu M, Chang WJ. 2016. Chloroplast genomes: diversity, evolution, and applications in genetic engineering. Genome Biol. 17(1):134.
- Dierckxsens N, Mardulyn P, Smits G. 2017. NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 45(4):e18.
- Doležalová I, Lebeda A, Janeček J, Číhalíková J, Křístková E, Vránová O. 2002. Variation in chromosome numbers and nuclear DNA contents in genetic resources of Lactuca L. species (Asteraceae). Genet Resour Crop Evol. 49(4):385–397.
- Frazer KA, Pachter L, Poliakov A, Rubin EM, Dubchak I. 2004. VISTA: computational tools for comparative genomics. Nucleic Acids Res. 32(Web Server issue):W273–W279.
- Guo X, Liu J, Hao G, Zhang L, Mao K, Wang X, Zhang D, Ma T, Hu Q, Al-Shehbaz IA, et al. 2017. Plastome phylogeny and early diversification of Brassicaceae. BMC Genomics . 18(1):176.
- iPlant. 2020a. Lactuca raddeana. [accessed 2020 Dec]. http://www.iplant.cn/info/Lactuca%20raddeana
- iPlant. 2020b. Lactuca. [accessed 2020 Dec]. http://www.iplant.cn/info/Lactuca?t=z
- Keeling PJ. 2004. Diversity and evolutionary history of plastids and their hosts. Am J Bot. 91(10):1481–1493.
- Kurtz S, Choudhuri JV, Ohlebusch E, Schleiermacher C, Stoye J, Giegerich R. 2001. REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 29(22):4633–4642.
- Langmead B, Trapnell C, Pop M, Salzberg SL. 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10(3):R25.
- Lebeda A, Astley D, Kristkova E. World genetic resources of Lactuca spp.: their taxonomy and biodiversity. In: Lebeda A, Křístková E, editors. Eucarpia leafy vegetables ’99. Olomouc (Czech Republic): Palacký University; 1999; p. 81–94.
- Li SY, Sun J, Niu BX, Yan FL, Zhan HQ. 2011. Effect of lactuside B on the expression of bcl-2 and bax mRNA and their protein in rats' cerebral cortex after cerebral ischemia-reperfusion injury. Yao Xue Xue Bao = Acta Pharmaceutica Sinica. 46:1314–1320.
- Li X-W, Hu Z-G, Lin X-H, Li Q, Gao H-H, Luo G-A, Chen S-L. 2012. High-throughput pyrosequencing of the complete chloroplast genome of Magnolia officinalis and its application in species identification. Yao Xue Xue Bao. 47(1):124–130.
- Lu H, Redus MA, Coburn JR, Rutger JN, McCouch SR, Tai TH. 2005. Population structure and breeding patterns of 145 US rice cultivars based on SSR marker analysis. Crop Sci. 45(1):cropsci2005.0066–76.
- Misra S, Harris, N. 2006. Using Apollo to browse and edit genome annotations. Curr Protoc Bioinformatics. Chapter 9:Unit 9.5.
- Palmer JD. 1991. Plastid chromosomes: structure and evolution. Mol Biol Plast. 7:5–53.
- Rice P, Longden I, Bleasby A. 2000. EMBOSS: the European molecular biology open software suite. Trends Genet. 16(6):276–277.
- Shaw J, Lickey EB, Schilling EE, Small RL. 2007. Comparison of whole chloroplast genome sequences to choose noncoding regions for phylogenetic studies in angiosperms: the tortoise and the hare III. Am J Bot. 94(3):275–288.
- Shi L, Chen H, Jiang M, Wang L, Wu X, Huang L, Liu C. 2019. CPGAVAS2, an integrated plastome sequence annotator and analyzer. Nucleic Acids Res. 47(W1):W65–W73.
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30(9):1312–1313.
- Tanaka K, Ohtake R, Yoshida S, Shinohara T. 2017. Effective DNA fragmentation technique for simple sequence repeat detection with a microsatellite-enriched library and high-throughput sequencing. BioTechniques. 62(4):180–182.
- Thompson JD, Gibson TJ, Higgins DG. 2002. Multiple sequence alignment using ClustalW and ClustalX. Curr Protoc Bioinformatics. Chapter 2: Unit 2.3.
- Wang ZH, Peng H, Kilian N. 2013. Molecular phylogeny of the Lactuca alliance (Cichorieae subtribe Lactucinae, Asteraceae) with focus on their Chinese centre of diversity detects potential events of reticulation and chloroplast capture. PLoS One. 8(12):e82692.
- Wei Z, Zhu S-X, Van den Berg RG, Bakker FT, Schranz ME. 2017. Phylogenetic relationships within Lactuca L. (Asteraceae), including African species, based on chloroplast DNA sequence comparisons. Genet Resour Crop Evol. 64(1):55–71.
- Wei Z. 2016. Genetic diversity and evolution in Lactuca L. (Asteraceae): from phylogeny to molecular breeding. Wageningen: Wageningen University.