
Abstract
The complete chloroplast genome of Sibbaldianthe bifurca Linnaeus was sequenced, assembled and annotated. It is a circular form of 156,734 bp in length, which was separated into four distinct regions, a large single copy (LSC) of 83,450 bp, a small single-copy region (SSC) of 18,286 bp, two inverted repeats (IR) of 27,499 bp. After annotation, a total of 133 genes were predicted, of which, 87 encode proteins, eight rRNA, 38 tRNA. The evolutionary history, inferred using the Maximum Likelihood (ML) method, indicates that S. bifurca was grouped within Rosaceae, and comprised a clade with Sibbaldianthe adpressa with 100% Bootstrap value.
Sibbaldianthe bifurca Linnaeus, belonging to Rosaceae, is a sympodially growing long-rooted, nonrosette, polycarpic perennial herb with yellow flowers, and has a wide range of distribution, from European Russia, West Siberia, Central Asia Mongolia to China (Basargin Citation2007; Godin Citation2009). For containing some chemical constituents, such as flavonoid, this plant was used extensively in traditional medicines for curing ulceration, cancer, hepatitis, etc., and some bioactive constituents have been identified, such as quercetin-4′-O-beta-D-glucoside from methanol extracts of this plant. It inhibits tyrosinase strongly (Zhao et al. Citation2008; Piao et al. Citation2009; Tomczyk et al. Citation2010). Apart from researches on medicines, some literatures on the population structure and ontogeny have also been reported (Basargin Citation2007; Godin Citation2009). In this study, we report the complete chloroplast (cp) genome of S. bifurca.
Samples from Qilian mountains (36°34’37"N,101°48’27"E) in Qinghai Province were collected for sequencing which was performed on the Illumina Novaseq platform (Shenzhen Huitong biotechnology Co. Ltd). Voucher specimen (HCPQNU-20200531001) was deposited in the Herbarium, College of Pharmacy, Qinghai Nationalities University. A sample’s total genomic DNA was extracted from about 100 mg fresh leaves using a modified CTAB method (Murray and Thompson Citation1980). Paired-end Libraries with an average length of 350 bp were constructed and sequenced. The complete cp genome was assembled with the de novo assembler SPAdes (Bankevich et al. Citation2012) and annotated via PGA (Qu et al. Citation2019).
The complete cp genome of S. bifurca (GenBank accession no. MT796626.1) has a typical quadripartite form of 156,734 bp in length and composed of a large single-copy region (LSC, 83,450 bp), a small single-copy region (SSC, 18,286 bp), two inverted repeats (IR, 27,499 bp). GC content of the genome is 37.1%. A total of 133 genes were predicted on this cp genome, of which, 87 encode proteins, eight rRNA, and 38 tRNA.
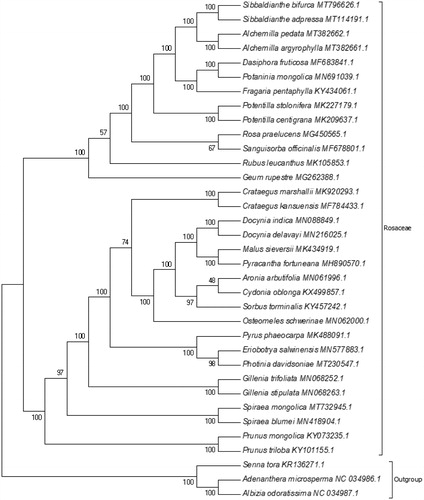
Phylogenetic analysis was performed based on complete cp genomes of S. bifurca and other 31 related species reported in Rosaceae, three species in Leguminosae as out-group. The sequences were aligned using MAFFT (Katoh et al. Citation2002), and trimAl was employed to remove ambiguously aligned sites (Capella-Gutierrez et al. Citation2009). The evolutionary history was inferred with the Maximum Likelihood (ML) method using IQ-TREE 1.6.12 under GTR + F+R3 model (Nguyen et al. Citation2015; Kalyaanamoorthy et al. Citation2017), and the output file was edited in MEGA 7.0 (Kumar et al. Citation2016). Bootstrap (BS) values were calculated with UFBoot2 from 1000 replicates analysis (Hoang et al. Citation2017). As expected, S. bifurca was grouped within Rosaceae and comprised a clade with Sibbaldianthe adpressa with 100% BS value (). The complete cp genome of S. bifurca will be helpful for further studies on population genetics, taxonomy or resources protection.
Figure 1. ML phylogenetic tree based on 35 species chloroplast genomes was constructed using IQ-TREE 1.6.12. Numbers on each node are bootstrap from 1000 replicates.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The genome sequence data that support the findings of this study are openly available in GenBank of NCBI at (https://www.ncbi.nlm.nih.gov/nuccore/MT796626.1) under the accession no. MT796626.1. The associated BioProject, SRA, and Bio-Sample numbers are PRJNA678812, SRR13070755, and SAMN16813076, respectively.
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article.
Additional information
Funding
References
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, Pyshkin AV, et al. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 19(5):455–477.
- Basargin EA. 2007. Ecological–demographic characterization of Potentilla bifurca L. cenopopulations. Russ J Ecol. 38(5):323–326.
- Capella-Gutierrez S, Silla-Martinez JM, Gabaldon T. 2009. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics. 25(15):1972–1973.
- Hoang DT, Chernomor O, von HA, Minh BQ, Vinh LS. 2017. UFBoot2: improving the ultrafast bootstrap approximation. Mol Biol Evol. 35(2):518–522.
- Godin VN. 2009. Population structure of Potentilla bifurca L. in the Altai–Sayan Mountain Region. Contemp Probl Ecol. 2(5):456–461.
- Kalyaanamoorthy S, Minh BQ, Wong TKF, von Haeseler A, Jermiin LS. 2017. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods. 14(6):587–589.
- Katoh K, Misawa K, Kuma K, Miyata T. 2002. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30(14):3059–3066.
- Kumar S, Stecher G, Tamura K. 2016. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 33(7):1870–1874.
- Murray MG, Thompson WF. 1980. Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res. 8(19):4321–4326.
- Nguyen LT, Schmidt HA, von Haeseler A, Minh BQ. 2015. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 32(1):268–274.
- Tomczyk M, Pleszczyńska M, Wiater A. 2010. Variation in total polyphenolics contents of aerial parts of Potentilla species and their anticariogenic activity. Molecules. 15(7):4639–4651.
- Piao XL, Tian YZ, Mi XY, Cui J. 2009. Tyrosinase inhibition of Potentilla bifurca. Zhongguo Zhong Yao Za Zhi. 34(15):1952–1954.
- Qu XJ, Moore MJ, Li DZ, Yi TS. 2019. PGA: a software package for rapid, accurate, and flexible batch annotation of plastomes. Plant Methods. 15:50.
- Zhao YL, Cai GM, Hong X, Shan LM, Xiao XH. 2008. Anti-hepatitis B virus activities of triterpenoid saponin compound from Potentilla anserine L. Phytomedicine. 15(4):253–258.