
Abstract
The present study for the first time describes the complete mitochondrial (mt) genome of Antheraea pernyi Guérin-Méneville 1855 strain Luhong, a genetic lethal mutant exhibiting especially red skin color. The mt genome is 15,563 bp in length that is the smallest among the sequenced A. pernyi inbred strains. This genome displays an identical genomic component and gene order to other six known A. pernyi mt genomes. The mt genome-based phylogenetic analysis clustered Luhong with four strains exhibiting yellow skin color, consistent with the traditional view that all of them belonged to the yellow blood lineage.
Chinese oak silkmoth, Antheraea pernyi Guérin-Méneville 1855 is not only an edible insect for human food but also a traditional silk-producing insect. It has already been used as an ornamental insect for agricultural tourism in China due to its colorful larvae including yellow-cyan, white, blue, and yellow. Up to now, more than 100 inbred strains with diverse agronomic characters have been developed. Luhong is one such strain exhibiting especially red skin color used for agricultural tourism. The homozygotes of this strain (∼25%) are genetically lethal during embryonic development. The remaining individuals exhibit yellow skin color from 2nd to 4th instar stages, but in the 5th instar stage two-thirds become red and one-thirds maintain yellow (Liu et al. Citation1999). The founder individuals of this strain with red skin color was found from a local strain ‘Luhuang_1’ exhibiting yellow skin color in Lushan of Henan Province (Sericultural Institute of Liaoning Province Citation1994). The present study determines the mitochondrial (mt) genome of this strain for the first time to understand the basic genetic information.
Specimens of the inbred strain Luhong were provided by the Sericultural Institute of Henan Province (N33°26′; E112°44′), Zhengzhou, China. Specimens used in this study were preserved in 95% ethanol and deposited at the Department of Sericulture, Shenyang Agricultural University (Yan-Qun Liu, [email protected]) under the voucher number OAK_SILKWORM_LUHONG_01. The total genomic DNA was extracted from a single individual. The long PCR amplification, sequence determination and genomic annotation were done as previously described for Qinghuang_1 (Li et al. Citation2020). The mt genome was manually annotated by comparing with the published mt genomes of A. pernyi.
The full mt genome of strain Luhong is 15,563 bp in length and also contains a typical insect mt gene set including 13 protein-coding genes (PCGs), 22 tRNA genes, and two ribosomal RNA genes. The genomic component and gene order are identical to other six known A. pernyi mt genomes as well as known Bombycidea species. The A + T-rich region is 552 bp in size harboring an 18 bp poly-T stretch preceded by the ATAGA motif but a 19 bp poly-T stretch in the other six known A. pernyi mt genomes. In the A + T-rich region, a 16 bp poly-A element upstream tRNAMet and intermittently interrupted by T and G is also present for Luhong; however, a 16 bp poly-A element intermittently interrupted by G occurs in two strains (Yu_7 and 731) and a 15 bp poly-A element intermittently interrupted by G occurs in four strains (Yu_6, Yuzao_1, Qing_6 and Qinghuang_1). Comparison analysis indicated that length variation of the two conserved structural features has occurred among strains of A. pernyi.
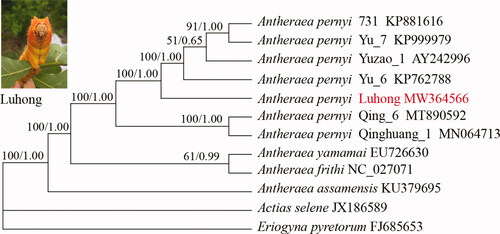
The intraspecific phylogenetic relationships of seven A. pernyi strains available to date were assessed based on the nucleotide sequence of mt genomes with maximum-likelihood and Bayesian inference methods under GTR + G + I nucleotide substitution model (). Three Antheraea species A. yamamai, A. frithi, and A. assamensis were also included and two non-Antheraea species served as outgroups. Bayesian inference and maximum-likelihood methods were conducted using MrBayes ver. 3.1.2 and IQ-TREE 1.6.12, respectively (Huelsenbeck and Ronquist Citation2001; Nguyen et al. Citation2015). The two phylogenetic trees obtained are congruent with strong support for almost all nodes. The phylogenetic analyses recovered two groups for the seven A. pernyi strains used. Group one contained Qing_6 and Qinghuang_1, both of them displaying yellow-cyan skin color were derived from ‘Aiyang’ population of Liaoning Province. Another group contained five strains, all of which showing yellow skin color were derived from ‘Lushan’ population of Henan Province. The mt genome-based phylogenetic analysis clustered Luhong with the other four strains with yellow skin color, consistent with their breeding histories.
Figure 1. Phylogenetic trees inferred from full mitochondrial genomes using maximum-likelihood and Bayesian inference methods under GTR + G + I model. The bootstrap values (former) and posterior probability (latter) values are indicated at the nodes. GenBank accession numbers are listed following the name of each species or strain.
Disclosure statement
The authors declare no competing financial interest. The authors alone are responsible for the content and writing of the paper.
Data availability statement
The genome sequence data that support the findings of this study are openly available in GenBank of NCBI at (https://www.ncbi.nlm.nih.gov/) under the accession no. MW364566. The associated BioProject, SRA, and Bio-Sample numbers are PRJNA695117, SRR13558698, and SAMN17601314, respectively.
Additional information
Funding
References
- Huelsenbeck JP, Ronquist F. 2001. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics. 17(8):754–755.
- Li XY, Liu YC, Zhang RS, Chen DB, Chen MM, Li YP, Liu YQ, Qin L. 2020. The mitochondrial genome of Qinghuang_1, the first modern improved strain of Chinese oak silkworm, Antheraea pernyi (Lepidoptera: Saturniidae). J Insect Food Feed. 7(2):233–243. https://doi.org/doi.10.3920/JIFF2020.0054.
- Liu ZG, Wang SF, Zhang XY. 1999. Genetic analyses on the larva skin colors of Qing 6, Luhong and Xiaoxinghuang Race in Chinese tusser. J Shenyang Normal Univ. 2:37–43.
- Nguyen LT, Schmidt HA, von Haeseler A, Minh BQ. 2015. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 32(1):268–274.
- Sericultural Institute of Liaoning Province, ed. 1994. The records of tussah varieties in China. Shenyang (China): Liaoning Technology Press; p. 1–274.