
Abstract
Thalassiosira is a species-rich genus with about 170 described species, many of which are harmful algal species with significant negative ecological impact. However, genome data of these species remain limited. In this study, the complete mitochondrial genome of Thalassiosira profunda (Hendey) Hasle 1973 was determined for the first time. The circular genome was 40,470 bp in length with GC content of 30.98%. It encodes 63 genes including 36 protein-coding genes (PCGs), 25 tRNA genes, and two rRNA genes. Phylogenetic analysis using concatenated PCGs suggested that T. profunda had a closer evolutionary relationship with Skeletonema marinoi of a different family (Skeletonemataceae) than Thalassiosira pseudonana, suggesting complex evolutionary relationship among species in these two families. Colinearity analysis also revealed fewer genome rearrangements between T. profunda and S. marinoi than that between T. profunda and T. pseudonana. This study suggests that mitochondrial genomes of many more species in the Thalassiosiraceae and Skeletonemataceae families are needed to disentangle the complex evolutionary relationships in the order of Thalassiosirales.
Thalassiosira (Mediophyceae, Bacillariophyta) is a species-rich genus with about 170 species described globally (Guiry and Guiry Citation2020) and about 50 species described in China (Li Citation2006). At least 10 Thalassiosira species, such as Thalassiosira rotula, Thalassiosira diporocyclus, and Thalassiosira weissflogii, have been found to form blooms with negative impact on environment (Li Citation2006; Li et al. Citation2013). Despite their important role in environment and ecology, molecular analysis of species in this genus has been limited. Here, we constructed the complete mitochondrial genome of Thalassiosira profunda (Hendey) Hasle 1973. The strain CNS00050 was isolated in water samples collected during an expedition to the Jiaozhou Bay (36°01.481′N, 120°17.202′E) in March 2019 onboard the research vehicle ‘Innovation’. The strain CNS00050 was confirmed to be T. profunda based on its morphological features and molecular sequences. The cells of CNS00050 were small, with diameters being 3–5 µm. Phylogenetic analysis of full-length 18S rDNA sequences indicated that the full-length 18S rDNA sequence of CNS00050 (MW205689) clustered with four T. profunda 18S rDNA sequences (KC284713, MN528652, MN528651, and MN528654) reported previously (Alverson Citation2016; Arsenieff et al. Citation2020). Another T. profunda full-length 18S rDNA sequence (AM235383) was clustered with Thalassiosira nordenskioeldii. However, this sequence was not supported by any published evidence. Similar phylogenetic analysis of other molecular markers including 28S rDNA D1-D2 regions (MW205747), rbcL (MW478286), and ITS (MW474850) all supported that the strain CNS00050 was T. profunda. Its specimen was deposited in the collection of marine algae in KLMEES of IOCAS (Nansheng Chen, [email protected]) under the voucher number CNS00050.
Illumina sequencing results of T. profunda were assembled into scaffolds using SPAdes v3.13.2 (Bankevich et al. Citation2012) and Platanus-allee v2.2.2 (Kajitani et al. Citation2019). Scaffolds of target mitochondrial genomes were selected from the assembly results using BLASTN v2.10.0. The mitochondrial genome sequence was examined using DOTTER v4.44.1 (Sonnhammer and Durbin Citation1995) and validated using the MEM algorithm of BWA v0.7.17 (Li and Durbin Citation2010). The alignments were visualized using IGV v2.8.12 (Robinson et al. Citation2011). Open reading frames (orfs) in the mitochondrial genome were first identified using Open Reading Frame Finder (ORF finder) (https://www.ncbi.nlm.nih.gov/orffinder) with ‘Genetic code: 4, ORF start codon: ‘ATG’ only’ selected. Protein-coding genes (PCGs) annotation was performed by using SmartBLAST (https://blast.ncbi.nlm.nih.gov/smartblast/) and BLASTP. tRNA genes were annotated using tRNAscan-SE 2.0 (Chan and Lowe Citation2019) with default setting. The locations of rRNAs were predicted by MFannot (https://megasun.bch.umontreal.ca/RNAweasel/) and determined by direct alignment with the mitogenomes of related species using MEGA X (Kumar et al. Citation2018) and BLASTN. The annotations were converted into genome maps by using OrganellarGenomeDRAW (OGDRAW) (Greiner et al. Citation2019).
The complete mitochondrial genome of T. profunda (GenBank accession number: MW013551) is 40,470 bp in size with GC content of 30.98%. It encodes 63 genes including 36 PCGs, 25 tRNA genes, and two rRNA genes. Among the 36 PCGs, 34 genes start with the canonical ATG start codons, nad11 with TTG, and atp8 with ATT. Most genes have canonical stop codons TAA (31 of 36 genes), with five genes having TAG as stop codons. The 25 tRNA genes, ranging in length from 72 bp to 89 bp, have typical cloverleaf secondary structures. No introns were found in the T. profunda mitochondrial genome.
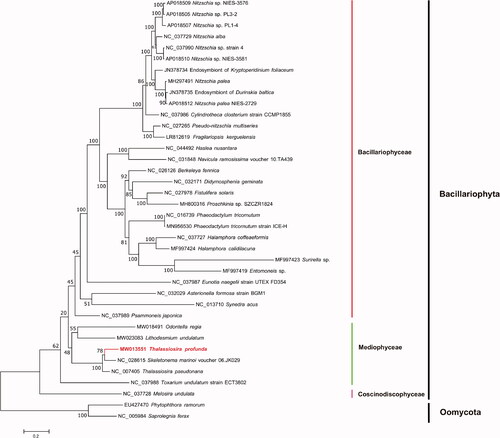
Maximum-likelihood (ML) phylogenetic tree () was constructed using tandem amino acid sequences of 31 common genes including atp6, 8, 9; cob; cox1, 2, 3; nad1–7, 4L, 9, 11; rpl2, 5, 6, 14, 16; rps3, 4, 8, 10, 11, 13, 14, 19; and tatC, from 35 publicly diatom mitochondrial genomes using IQtree v1.6.12 (Trifinopoulos et al. Citation2016) with 1000 bootstrap alignments. Mitochondrial genomes of two Oomycota species Phytophthora ramorum (EU427470) and Saprolegnia ferax (NC_005984) were used as out-group taxa. The results demonstrated that species fell nicely into three clades corresponding to three classes of the phylum Bacillariophyta including Coscinodiscophyceae, Mediophyceae, and Bacillariophyceae. T. profunda was grouped with Skeletonema marinoi and T. pseudonana with strong support. T. profunda of the family Thalassiosiraceae showed closer evolutionary relationship with S. marinoi of the family Skeletonemataceae than that with T. pseudonana, which were different families in the order Thalassiosirales (Stoermer Citation2003). Furthermore, colinearity analysis of the mitochondrial genomes of three species T. profunda, T. pseudonana, and S. marinoi identified a single inversion event involving a single gene atp6 between T. profunda and S. marinoi, while identified an inversion event involving atp6 plus a translocation event involving two genes cox2-cox3 between T. profunda and T. pseudonana, also suggesting higher similarity between T. profunda and S. marinoi. These results were consistent with findings from a recent study suggesting that T. pseudonana should be classified as a species of another genus Cyclotella (Alverson et al. Citation2011). Thus, the mitochondrial genome of T. profunda likely represents that first mitochondrial genome of Thalassiosira. The complete mitochondrial genomes of more species in Thalassiosira and related genus will help to clarify the evolutionary relationships and classification of the order Thalassiosirales.
Figure 1. Maximum-likelihood (ML) phylogenetic tree based on tandem amino acid sequences of 31 common genes from 35 publicly diatom mitochondrial genomes, and Phytophthora ramorum (EU427470) and Saprolegnia ferax (NC_005984) in Oomycota were used as out-group taxa. The numbers beside branch nodes are the percentage of 1000 bootstrap values.
Acknowledgements
We are thankful to all staffs of marine ecological environment genomics research group in Institute of Oceanology, Chinese Academy of Sciences. We are grateful to colleagues from the Jiaozhou Bay Marine Ecosystem Research Station for their help in field sampling.
Disclosure statement
The authors are responsible for the content and writing of the paper. The authors report no conflicts of interest.
Data availability statement
The genome sequence data that support the findings of this study are openly available in GenBank of NCBI at https://www.ncbi.nlm.nih.gov/nuccore/MW013551, under the accession no. MW013551. The associated BioProject, SRA, and Bio-Sample numbers are PRJNA684688 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA684688), SRR13245496 (https://www.ncbi.nlm.nih.gov/sra/SRR13245496), and SAMN17065834 (https://www.ncbi.nlm.nih.gov/biosample/SAMN17065834/), respectively.
Additional information
Funding
References
- Alverson AJ. 2016. Timing marine–freshwater transitions in the diatom order Thalassiosirales. Paleobiology. 40:91–101.
- Alverson AJ, Beszteri B, Julius ML, Theriot EC. 2011. The model marine diatom Thalassiosira pseudonana likely descended from a freshwater ancestor in the genus Cyclotella. BMC Evol Biol. 11:125.
- Arsenieff L, Le Gall F, Rigaut-Jalabert F, Mahe F, Sarno D, Gouhier L, Baudoux AC, Simon N. 2020. Diversity and dynamics of relevant nanoplanktonic diatoms in the Western English Channel. ISME J. 14:1966–1981.
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, et al. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 19:455–477.
- Chan PP, Lowe TM. 2019. tRNAscan-SE: searching for tRNA genes in genomic sequences. Methods Mol Biol. 1962:1–14.
- Greiner S, Lehwark P, Bock R. 2019. OrganellarGenomeDRAW (OGDRAW) version 1.3.1: expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 47:W59–W64.
- Guiry MD, Guiry GM. 2020. AlgaeBase. World-wide electronic publication. Galway: National University of Ireland. http://www.algaebase.org.
- Kajitani R, Yoshimura D, Okuno M, Minakuchi Y, Kagoshima H, Fujiyama A, Kubokawa K, Kohara Y, Toyoda A, Itoh T. 2019. Platanus-allee is a de novo haplotype assembler enabling a comprehensive access to divergent heterozygous regions. Nat Commun. 10:1702.
- Kumar S, Stecher G, Li M, Knyaz C, Tamura K. 2018. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 35:1547–1549.
- Li H, Durbin R. 2010. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics. 26:589–595.
- Li Y. 2006. Taxonomic studies and ecological characteristics on Nano-diatoms in coastal waters of China. Xiamen University, Xiamen, China.
- Li Y, Zhao Q, Lü S. 2013. The genus Thalassiosira off the Guangdong coast, South China Sea. Bot Mar. 56:83–110.
- Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. 2011. Integrative genomics viewer. Nat Biotechnol. 29:24–26.
- Sonnhammer ELL, Durbin R. 1995. A dot-matrix program with dynamic threshold control suited for genomic DNA and protein sequence analysis. Gene. 167:GC1-GC10.
- Stoermer EF, Julius ML. 2003. Centric Diatoms. In: Freshwater Algae of North America. (Wehr, J.D. & Sheath, R.G. Eds), pp. 559-594. San Diego: Academic Press.
- Trifinopoulos J, Nguyen LT, von Haeseler A, Minh BQ. 2016. W-IQ-TREE: a fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 44:W232–W235.