
Abstract
The complete mitochondrial DNA sequence of the Japanese field vole Microtus montebelli was determined using Illumina MiSeq platform. The assembled genome was 16,307 bp in length and contained 13 protein-coding genes, two ribosomal RNA genes, 22 transfer RNA genes. According to phylogenetic analysis of 13 protein-coding genes, M. montebelli and other Microtus species consist of paraphyletic clades and M. montebelli is most closely related to M. kikuchii, a species endemic to Taiwan.
The Japanese field vole Microtus montebelli is endemic to Japan, inhabiting cultivated fields, meadows, conifer plantations, and riverbanks (Ohdachi et al. 2015). Although they have been recognized as a pest that eats the bark and roots of apple orchard trees in areas with heavy snowfall, they are a locally endangered species in Kyushu and urban areas in Honshu (Murano et al. Citation2019). This warrants the development of genetic markers with varying evolutionary rates to infer dispersal patterns and genetic population structure of M. montebelli.
The specimen of M. montebelli was collected from an apple orchard in Hirosaki, Japan (40°34′48″N, 140°25′18″E) on 13 May 2019, with permission from Aomori Prefecture (order number: 4153), following Hirosaki University guidelines for the ethical treatment of animals. The specimen was deposited in Hirosaki University (Dr. Atsushi Sogabe, e-mail: [email protected]) under the voucher number HUA1900522. The genomic DNA was extracted using a DNeasy Tissue and Blood Kit (Qiagen, Hilden, Germany). Whole-genome shotgun sequencing (2 × 300 bp) was performed using the Illumina MiSeq platform (Illumina, Hayward, CA), yielding 7,798,791 paired raw reads. Assembly was conducted via MITObim v1.9 (Hahn et al. Citation2013) using M. arvalis (GenBank accession number: MG_948434) as the reference. The assembled mitogenome sequence was annotated using the MITOS (Bernt et al. Citation2013).
The complete mitogenome of M. montebelli was 16,307 bp in length and contained 13 protein-coding genes (PCGs), two rRNA genes, 22 tRNA genes, one origin of L strand replication, and one control region. The gene arrangement of M. motebelli was identical to that of other species in Arvicolinae. All PCGs initiate with ATN codons (ND1 began with ATA, and ND2, ND3 and ND5 began with ATT). TAA and TAG are the termination codon for most of the PCGs, whereas the incomplete stop codons (TA–) were used in ND4 and COX3. The overall nucleotide composition was A (32.8%), C (27.5%), G (13.3%), and T (26.4%), indicating an obvious A/T skew (59.2%).
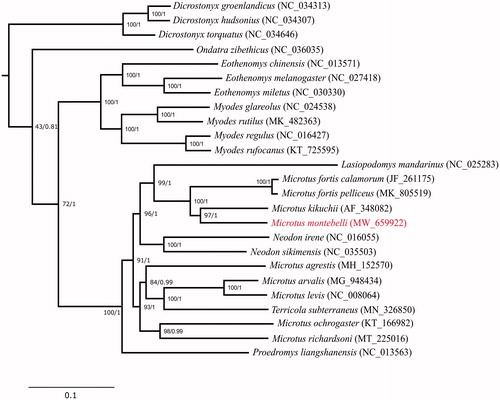
Bayesian inference (BI) and maximum-likelihood (ML) were used to reconstruct the phylogenetic trees based on 13 protein-coding genes from 25 species of Arvicolinae with the Chinese hamster Cricetulus griseus as an outgroup. Phylogenetic analysis was conducted with BEAST 1.10.4 (Suchard et al. Citation2018) and RAxML-NG 1.0.1 (Kozlov et al Citation2019) for BI and ML, respectively. BI and ML yielded identical phylogenetic trees (). Results of phylogenetic tree analysis showed that genus Microtus are paraphyletic, sharing a common ancestor with genus Lasiopodomys and Neodon. Interestingly, we also identified that M. montebelli is a sister species of M. kikuchii, a species endemic to Taiwan.
Figure 1. Maximum-likelihood tree of the subfamily Arvicolinae based on the sequences of 13 protein-coding genes (the outgroup is not shown for graphical reason). Number beside each node indicates bootstrap support values for ML (left) and posterior probabilities support values for BI (right).
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The data that support the findings of this study are available in NCBI at https://www.ncbi.nlm.nih.gov/, reference number MW659922.
References
- Bernt M, Donath A, Jühling F, Externbrink F, Florentz C, Fritzsch G, Pütz J, Middendorf M, Stadler PF. 2013. MITOS: improved de novo metazoan mitochondrial genome annotation. Mol Phylogenet Ecol. 69(2):313–319.
- Hahn C, Bachmann L, Chevreux B. 2013. Reconstructing mitochondrial genomes directly from genomic next-generation sequencing reads – a baiting and iterative mapping approach. Nucleic Acids Res. 41(13):e129–e129.
- Kozlov AM, Darriba D, Flouri T, Morel B, Stamatakis A. 2019. RAxML-NG: a fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics. 35(21):4453–4455.
- Murano C, Kasahara S, Kudo S, Inada A, Sato S, Watanabe K, Azuma N. 2019. Effectiveness of vole control by owls in apple orchards. J Appl Ecol. 56(3):677–687.
- Ohdachi S, Ishibashi Y, Iwasa M, Fukui D, Saitoh T. 2015. The wild mammals of Japan. 2nd ed. Kyoto: Shokado.
- Suchard MA, Lemey P, Baele G, Ayres DL, Drummond AJ, Rambaut A. 2018. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Vir Evol. 4:vey016.