
Abstract
The complete plastome of Pinellia peltata is reported in this study. The whole plstome contains 164,293 bp, including a large single copy region (90,089 bp) and a small single copy region (24,871 bp), which were separated by a pair of inverted repeat regions (24,881 bp of IRB and 24,982 bp of IRA). Totally, 130 genes were identified, containing 86 coding-protein, 8 rRNA (4 rRNA species), and 36 tRNA genes. Phylogenetic analysis based on common protein coding genes of 10 Araceae species showed that P. peltata is original than other species in the genus Pinellia. The whole plastome of P. peltata and other species should facilitate further works for the genus Pinellia about genetic diversity, phylogenetic analysis, and so on.
Pinellia peltata Pei (Araceae) is a relative species of P. ternata, which is an important Chinese Traditional Medicine. P. peltata is distinguished from other relatives in the genus Pinellia by the peltate leaf blade and development of stem tuber (Zhu et al. Citation2007). Individuals of P. peltata used to sequencing are collected from wild (Wenzhou, Zhejiang Province, China) and cultured in laboratory (Guizhou University, Guiyang, Guizhou Province, China).
Total DNA were extracted from young leaves of one individual using a modified CTAB method (Doyle and Doyle Citation1987) and sequenced by Illumina NovaSeq Platform (Illumina, San Diego, CA) with paired-end mode. Totally, 31,530,954 reads (4,729,643,100 bp) were obtained. After removal of adapter sequences by AdapterRemoval v2 (Schubert et al. Citation2016), high-quality clean reads that were corrected by SOAPec v2.01 (Luo et al. Citation2012) with default settings were initially mapped to plastome of P. ternata (KR270823) using BWA (Li and Durbin Citation2009) and SAMtools (Li and Durbin Citation2009). The assembly was executed with A5-miseq v20150522 (Coil et al, Citation2015) and SPAdes v3.9.0 (Bankevich et al. Citation2012) and resulted in a 164,923 bp scaffold. Subsequently, the plastome annotation was carried out with GeSeq (Tillich et al. Citation2017) and PGA (Qu et al. Citation2019). The complete sequence and annotations of P. peltata plastome were submitted to GenBank with the accession number MT819952. The sequencing reads are available in the NCBI Sequence Read Archive (SRA) database under the accession number SRR12560323.
The whole chloroplast genome of P. peltata was 164,293 bp, consisting of a large single copy region (90,089 bp), a small copy region (24,871 bp), and a pair of inverted repeat regions. One bp indel in rrn23 between two copies located in IRs resulted in 24,881 bp of IRB and 24,982 bp of IRA. The contents of A, T, C, and G are 31.3%, 32.2%, 18.3%, and 18.2%, respectively. In total, 130 genes were identified: 86 coding-protein, 8 rRNA (4 rRNA species), and 36 tRNA genes. Except for 17 genes (all 4 rRNA, 6 tRNA, and 7 protein coding genes) with a double copy, other 96 genes are a single copy. Among these identified genes, 11 single copy genes (atpF, ndhA, petB, petD, rpl16, rpoC1, rps16, trnG-UCC, trnL-UAA, trnK-UUU, trnV-UAC) and 5 double-copy genes (ndhB, rpl2, ycf68, trnA-UGC, trnI-GAU) consist of 2 exons, and 2 single copy genes (ycf3 and clpP) consist three exons. The double copy gene rps12 share one exon in the LSC region and possess specifically two exons that are located in IR regions.
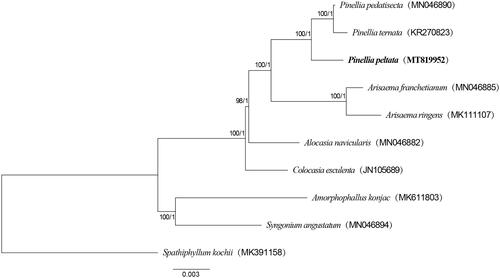
To validate systematic position of P. peltata, 10 species in Dracunculus clade of Araceae (Cusimano et al. Citation2011) were selected to phylogenetic analysis (Spathiphyllum kochii acted as outgroup). Seventy-eight common protein coding genes (PCGs, without repeat gene) from these plastomes were chosen to further analysis. Coding sequences of all PCGs were aligned using MAFFT v7.450 (Rozewicki et al. Citation2019) and conserved domains were identified using GBLOCKS 0.91 b (Talavera and Castresana Citation2007) with default parameters. Conserved sequences for all PCGs were then concatenated into a single sequence for each plastome. The phylogenetic relationships among the 10 Araceae species were determined by ML tree and BI tree were made by PhyloSuite v1.2.1 (Zhang et al. Citation2020). The phylogenetic tree () showed that three species of Pinellia and two species of Arisaema located in one branch, respectively. These results also indicated that whole plastome sequences are useful for population genetic and phylogenetic studies of species in Pinellia.
Figure 1. The phylogenetic tree of P. peltata and other species based on concatenated sequences that derived from 78 common protein coding genes (PCGs). The numbers on each node represent ML bootstrap values (1000 replicates)/BI posterior probability.
Disclosure statement
No potential conflict of interest was reported by the authors.
Data availability statement
The data that support the findings of this study will be available in GenBank at https://www.ncbi.nlm.nih.gov/, Accession no. MT819952.
Additional information
Funding
References
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, Pyshkin AV, et al. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 19(5):455–477.
- Coil D, Jospin G, Darling AE. 2015. A5-miseq: an updated pipeline to assemble microbial genomes from Illumina MiSeq data. Bioinformatics. 31(4):587–589.
- Cusimano N, Bogner J, Mayo SJ, Boyce PC, Wong SY, Hesse M, Hetterscheid WL, Keating RC, French JC. 2011. Relationships within the Araceae: comparison of morphological patterns with molecular phylogenies. Am J Bot. 98(4):654–668.
- Doyle JJ, Doyle JL. 1987. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull. 19:11–15.
- Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 25(14):1754–1760.
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup 2009. The sequence alignment/map format and samtools. Bioinformatics. 25(16):2078–2079.
- Luo R, Liu B, Xie Y, Li Z, Huang W, Yuan J, He G, Chen Y, Pan Q, Liu Y, et al. 2012. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience. 1(1):18.
- Qu XJ, Moore MJ, Li DZ, Yi TS. 2019. PGA: a software package for rapid, accurate, and flexible batch annotation of plastomes. Plant Methods. 15:50.
- Rozewicki J, Li S, Amada KM, Standley DM, Katoh K. 2019. MAFFT-DASH: integrated protein sequence and structural alignment. Nucleic Acids Res. 47(W1):W5–W10.
- Schubert M, Lindgreen S, Orlando L. 2016. 2016. AdapterRemoval v2: rapid adapter trimming, identification, and read merging. BMC Res Notes. 9(1):88.
- Talavera G, Castresana J. 2007. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst Biol. 56(4):564–577.
- Tillich M, Lehwark P, Pellizzer T, Ulbricht-Jones ES, Fischer A, Bock R, Greiner S. 2017. GeSeq – versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 45(W1):W6–W11.
- Zhang D, Gao F, Jakovlić I, Zou H, Zhang J, Li WX, Wang GT. 2020. PhyloSuite: an integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol Ecol Resour. 20(1):348–355.
- Zhu GG, Li H, Li R. 2007. A synopsis and a new species of the E Asian genus Pinellia (Araceae). Willdenowia. 37(2):503–522.