
Abstract
Haymondia wallichii DC. was a scandent shrub. In this study, we sequenced the complete chloroplast (cp) genome of H. wallichii to investigate its phylogenetic relationship in Fabaceae. The total length of the cp genome was 153,668 bp, consisted of a large single copy (LSC) region of 84,310 bp, a small single copy (SSC) region of 17,918 bp, and a pair of inverted repeat regions (IRs) of 25,720 bp. The genome contained 132 genes, namely 37 tRNA genes, 87 protein-coding genes, and 8 rRNA genes. The overall GC content was 35.4%. The phylogenetic analysis indicated H. wallichii was closely related to Pueraria montana var. thomsonii and Pueraria montana var. lobata.
Haymondia wallichii DC. is a species belonging to the family Fabaceae, which is mainly distributed in Yunnan and Tibet in China. H. wallichii is a common traditional Chinese medicine for anti-alcoholic and health care in Dali, Yunnan. In the previous study, H. wallichii was belonged to Pueraria (Egan and Pan Citation2015), while the classification of H. wallichii had always been controversial. Although Egan and Pan divided it into a new genus of Haymondia based on morphological characters and phylogenetic studies (Egan and Pan Citation2015; Egan et al. Citation2016), it was still incomplete in molecular biology research. The accurate identification of H. wallichii can ensure the quality of H. wallichii (Miao et al. Citation2019) and different molecular techniques may lead to different phylogenetic results (Zhang et al. Citation2020). Therefore, the study reported the complete chloroplast genome of the H. wallichii, and revealed its phylogenetic relationship with other Fabaceae plants to lay the foundation for future research of Haymondia.
In this study, The fresh leaves of H. wallichii were collected from Weishan mountain (N 25°53′71.57″, E 100°25′49.78″) of Dali, Yunnan province, China, and used as molecular materials. Meanwhile, voucher specimens with mature seeds were collected as the sample and deposited in the Herbarium of Medicinal Plants and Crude Drugs of the College of Pharmaceutical Science, Dali University (NO. XCL018). Total genomic DNA was extracted using the improved CTAB method (Doyle Citation1987) and sequenced by the Illumina Hiseq 2500 platform (Novogene, Tianjin, China). The raw data was filtered using Trimmomatic version 0.32 with default settings (Bolger et al. Citation2014). Then paired-end reads of clean data were assembled by GetOrganelle.py into circular contigs (Jin et al. Citation2018). The cp genome of H. wallichii was annotated in Geneious 9.1.4 (Kearse et al. Citation2012). A physical map of the cp genome was obtained through the web-based tool OGDraw version 1.2 (http://ogdraw.mpimp-golm.mpg.de/). Distributions of the simple sequence repeats (SSRs) were explored by the microsatellite search tool MISA (Thiel et al. Citation2003). The annotated cp genome sequences had been submitted to GenBank (accession number:MT797172.1).
The cp genome of H. wallichii was 153,668 bp in length, with 35.4% overall GC content, and exhibited a typical quadripartite structure, consisting of a large single copy (LSC) region of 84,310 bp, a small single-copy (SSC) region of 17,918 bp, and a pair of IRs (two inverted repeat regions) of 25,720 bp. It contained 132 genes, including 37 tRNA genes, 87 protein-coding genes, and 8 rRNA genes. A total of 88 simple sequence repeats (SSRs) with five types were detected in the cp genome, including 49 mononucleotide repeats, 28 dinucleotide repeats and 11 other types of SSR loci.
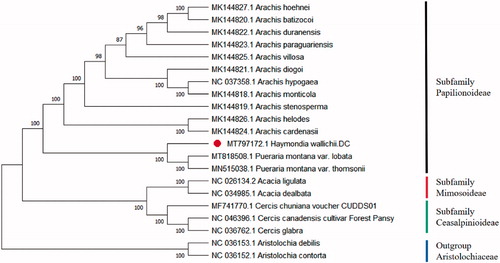
To reveal the phylogenetic relationship of the H. wallichii. A total of 18 cp genomes of Fabaceae were downloaded from NCBI database. The sequences were aligned by MAFFT v7.307 (Katoh and Standley Citation2013). Then the neighbour-joining (NJ) tree was established based on MEGA X (Kumar et al. Citation2018) and used Aristolochia of Aristolochiaceae as the outgroup (). Numbers in the nodes were the bootstrap values from 1000 replicates. The result showed that the cp genome of H. wallichii was closed to Pueraria montana var. thomsonii and Pueraria montana var. Lobata. This study of H. wallichii may provide a valuable guide to the Haymondia future researches of systematics and phylogenetic.
Figure 1. Phylogenetic position of H. wallichii inferred by the neighbour-joining (NJ) analysis based on 21 sequences, bootstrap values near the branch.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The data that supports the findings of this study is openly available in NCBI GenBank database at (https://www.ncbi.nlm.nih.gov/nuccore/MT797172.1) with the accession number is MT797172.1, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Additional information
Funding
References
- Bolger A, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 30(15):2114–2120.
- Doyle J. 1987. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull. 1(19):11–15.
- Egan AN, Pan B. 2015. Resolution of polyphyly in Pueraria (Leguminosae, Papilionoideae): The creation of two new genera, Haymondia and Toxicopueraria, the resurrection of Neustanthus, and a new combination in Teyleria. Phytotaxa. 3(218):201–226.
- Egan AN, Vatanparast M, Cagle W. 2016. Parsing polyphyletic Pueraria: delimiting distinct evolutionary lineages through phylogeny. Mol Phylogenet Evol. 104:44–59.
- Jin J, Yu W, Yang J, Song Y, Yi T, Li D. 2018. GetOrganelle: a simple and fast pipeline for de novo assembly of a complete circular chloroplast genome using genome skimming data. Genome Biol. 21(1):241.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30(4):772–780.
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, et al. 2012. Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 28(12):1647–1649.
- Kumar S, Stecher G, Li M, Knyaz C, Tamura K. 2018. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 35(6):1547–1549.
- Miao X, Niu J, Wang A, Wang D, Fan J. 2019. Complete chloroplast genome sequence of Pueraria thomsonii, an important traditional Chinese medicine plant. Mitochondr DNA B Resour. 2(4):4163–4165.
- Thiel T, Michalek W, Varshney R, Graner A. 2003. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor Appl Genet. 106(3):411–422.
- Zhang M, Pan B, Wang Y, Du X, Fu L, Zheng Y, Chen F, Wu W, Zhou Q, Ding S, et al. 2020. Recording the electrochemical profile of Pueraria leaves for polyphyly analysis. Chemistry Select. 17(5):5035–5040.