
Abstract
Herein, we report the complete chloroplast genome of Tilia mongolica Maxim. from Tiliaceae. The chloroplast genome of T. mongolica is 162,804 bp, with a large single copy region of 91,255 bp, small single copy region of 20,355 bp, and two inverted-repeat regions of 25,597 bp. The chloroplast genome contains 130 genes, including 85 protein-coding, 8 rRNA, and 37 tRNA. The total GC content is 36.46%. The phylogenetic analysis of T. mongolica showed a relatively close relationship with Tilia taishanensis.
The Tilia family includes approximately 50 genera and 450 species distributed worldwide. They are found throughout the temperate regions of America, Asia, and Europe (Fineschi et al. Citation2003). Tilia mongolica is a species of genus the Tilia. It is a deciduous tree, with light gray outer bark having obvious shallow grooves and cracks, and an inner bark rich in fiber and mucus. The leaves are mostly wide ovoid, with highly serrated edges. The flowers are yellow or white, and often occur in clusters. The fruit is round or oval nut-like, having one to three seeds. They are used as wood, honey resources, and ornamental trees (Cai et al. Citation2015; Boo and Park Citation2016). Traditionally, the lime tree has belonged to its own family, Tiliaceae (Aiello Citation1981). However, some molecular evidence has indicated that it is closely related to other plants (Bayer et al. Citation1999). Therefore, owing to limited taxonomic features and frequent hybridization, the classification of lime trees has been controversial (Pigott Citation2012). Previous studies on the genetic classification of Tilia populations had focused on the use of random amplified polymorphic DNA (Colagar et al. Citation2013; Filiz et al. Citation2015) and microsatellite (Logan et al. Citation2015) markers. With the development of sequencing technology, chloroplast genomes have been widely used for the identification and phylogenetic analyses of plant species, including some basswoods (Cai et al. Citation2015; Lu et al. Citation2020), owing to the highly conserved genome size and sequence (Sarzi et al. Citation2019; Sun et al. Citation2019). Therefore, it was imperative for the chloroplast genome sequence of T. mongolica to be resolved.
The fresh leaves of T. mongolica were collected from Beijing, China. The samples were stored in the Experimental Center of Forestry in North China (39.970°E, 116.096°N), Chinese Academy of Forestry. A specimen was deposited at the herbarium of Experimental Center of Forestry in North China (https://www.ncbi.nlm.nih.gov/nuccore/MW386998, contact person is Guangshun Zheng and email is [email protected]) under the voucher number mengduan-001. A CTAB protocol was used to isolate total genomic DNA (Wang et al. Citation2015). Sequencing libraries were generated using a TruSeq DNA Sample Preparation Kit (Illumina, USA) and a Template Prep Kit (Pacific Biosciences, USA). Genome sequencing was performed using the Pacific Biosciences and Illumina NovaSeq platforms. A total of 21,036,342 reads (including18,275,612 high-quality reads) were obtained, and the clean reads were assembled using SPAdes (Bankevich et al. Citation2012) and A5-miseq (Coil et al. Citation2015) to construct scaffolds and contigs. The chloroplast splicing results were obtained using these software packages, and the reference genome was analyzed using Mummerv3.1 software (Kurtz et al. Citation2004) to determine the positional relationships among and to fill the gaps between contigs. Results were corrected using Pilonv1.18 (Walker et al. Citation2014) software to obtain the final chloroplast genome. The assembled complete chloroplast genome sequence’s functional annotation was performed using the online program GeSeq (https://chlorobox.mpimp-golm.mpg.de/geseq.html). Related species were used as references, and the remaining parameters were set at default values.
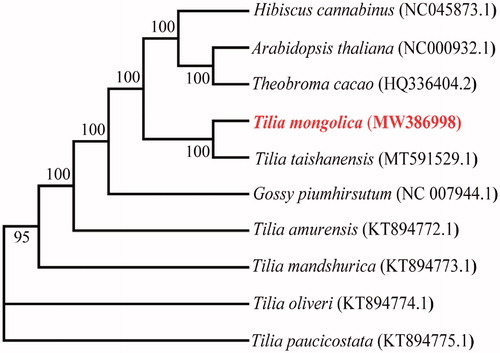
The chloroplast genome was annotated and submitted to GenBank (accession number MW386998). The chloroplast genome of T. mongolica is a typical circular DNA of 162,804 bp. It encodes two inverted repeat (IR) regions, IRa and IRb, with a large single copy (LSC) region, and a small single copy (SSC) region separating the IRs. The lengths of the LSC, SSC and IR regions are 91,255 bp, 20,355 bp, and 25,597 bp, respectively. The GC content of the chloroplast genome of T. mongolica is lower than the AT content, and the GC contents in the chloroplast genome, LSC, SSC, and IR regions are 36.46%, 34.06%, 31.05% and 42.90%, respectively. The chloroplast genome encodes 130 potentially functional genes, including 85 protein-coding, 8 rRNA, and 37 tRNA. rRNAs only exist in the IR region. In total, 16 genes are repeated in the IR region, 5 protein-coding, 7 tRNA, and 4 rRNA. Additionally, the ycf1 gene is at the junction between the IR and SSC, and rps19 is at the junction between the LSC and IR. In total, 16 genes (trnK, rps16, trnG, atpF, rpoC1, trnL-UAA, trnV-UAC, petB, petD, rpl16, rpl2, ndhB, rps12, trnL-GAU, trnA-UGC/UCG, and ndhA) contain one intron, and 2 genes (ycf3 and clpP1) contain two introns. The matK geneis located in trnK-UUU, and rps12 is a trans-spliced gene: the 5′-end is located in the LSC region, whereas the 3′-end is located in the two IR regions. The phylogenetic relationships of T. mongolica were estimated using the Maximum-likelihood method in MEGA7 (Kumar et al. Citation2016). The phylogenetic tree showed that T. mongolica is relatively closely ralated to the Tilia taishanensis ().
Figure 1. Phylogenetic tree constructed from 10 complete chloroplast genomes. The analysis was performed with the Maximum-likelihood using MEGA 7.0 software. The sequences were downloaded from NCBI GenBank, and the accession numbers are shown in parentheses.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The genome sequence data that support the findings of this study are openly available in GenBank of NCBI at [https://www.ncbi.nlm.nih.gov] (https://www.ncbi.nlm.nih.gov/) under the accession number MW386998. The associated BioProject, SRA, and Bio-Sample numbers are PRJNA701724, SRR13705393, and SAMN17905928, respectively.
Additional information
Funding
References
- Aiello A. 1981. An integrated system of classification of flowering plants. New York: Cambridge University Press.
- Bayer C, Fay MF, Bruijn AYD, Savolainen V, Morton CM, Kubitzki K, Alverson WS, Chase MW. 1999. Support for an expanded family concept of Malvaceae within a recircumscribed order Malvales: a combined analysis of plastid atpB and rbcL DNA sequences. Bot J Linn Soc. 129(4):267–303.
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, Pyshkin AV, et al. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 19(5):455–477.
- Boo D, Park SJ. 2016. Molecular phylogenetic study of Korean Tilia L. Korean J Plant Res. 29(5):547–554.
- Cai J, Ma PF, Li HT, Li DZ. 2015. Complete plastid genome sequencing of four Tilia species (Malvaceae): a comparative analysis and phylogenetic implications. PloS One. 10(11):e0142705.
- Coil D, Jospin G, Darling AE. 2015. A5-miseq: an updated pipeline to assemble microbial genomes from Illumina MiSeq data. Bioinformatics. 31(4):587–589.
- Colagar AH, Yusefi M, Zarei M, Yousefzadeh H. 2013. Assessment of genetic diversity of Tilia rubra DC. by RAPD analysis in the Hyrcanian forests, north of Iran. Pol J Ecol. 61:341–348.
- Filiz E, Birbilener S, Ozyigit II, Kulac S, Oruc FCS. 2015. Assessment of genetic variations of silver lime (Tilia tomentosa Moench.) by RAPD markers in urban and forest ecosystems. Biotechnol Biotechnol Equip. 29(4):631–636.
- Kumar S, Stecher G, Tamura K. 2016. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 33(7):1870–1874.
- Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, Antonescu C, Salzberg SL. 2004. Versatile and open software for comparing large genomes. Genome Biol. 5(2):R12.
- Logan SA, Phuekvilai P, Wolff K. 2015. Ancient woodlands in the limelight: delineation and genetic structure of ancient woodland species Tilia cordata and Tilia platyphyllos (Tiliaceae) in the UK. Tree Genet Genomes. 11(3):1–12.
- Lu YZ, Li WQ, Wang N, Wang Y, Han Y, Sun T, Liu LJ, Xie XM, Wang YH. 2020. Characterization of the complete chloroplast genome of Tilia taishanensis S. B. Liang (Tiliaceae). Mitochondrial DNA B Resour. 5(3):2719–2720.
- Fineschi S, Salvini D, Taurchini D, Carnevale S, Vendramin GG. 2003. Chloroplast DNA variation of Tilia cordata (Tiliaceae). Can J Res. 33(12):2503–2508.
- Pigott CD. 2012. Limes-tree and Basswoods a biological monograph of the genus Tilia. New York: Cambridge University Press.
- Sarzi DS, Haerolde L, Lopes FS, Furtado C, Oliveira DR, Sakuragui CM, Prosdocimi F. 2019. Complete plastid genome of Lippia origanoides (Verbenaceae) and phylogenomic analysis of Lamiales. Mitochondrial DNA Part B. 4(1):808–810.
- Sun Y, Yang H, Zhang Q, Qin L, Li P, Lee J, Chen S, Rahman K, Kang T, Jia M. 2019. Genetic diversity and its conservation implications of Vitex rotundifolia (Lamiaceae) populations in East Asia. Peer J. 7:e6194.
- Wang H, Pan G, Ma Q, Zhang J, Pei D. 2015. The genetic diversity and introgression of Juglans regia and Juglans sigillata in Tibet as revealed by SSR markers. Tree Genet Genomes. 11(1):1.
- Walker BJ, Abeel T, Shea T, Priest M, Abouelliel A, Sakthikumar S, Cuomo CA, Zeng Q, Wortman J, Young SK, et al. 2014. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PloS One. 9(11):e112963.