
Abstract
Nitraria sphaerocarpa Maxim. is a typical desert shrub commonly used as a sand binder. Here, we sequenced and characterized the whole plastid genome of N. sphaerocarpa. It is 159,369 bp in length, containing two copies of inverted repeat (IR) regions (26,566 bp, each), a large single-copy (LSC) region (87,854 bp), and a small single-copy (SSC) region (18,383 bp). It has 114 unigenes, including 79 protein-coding genes, 30 tRNA genes, four rRNA genes, and one pseudogene (infA). Phylogenetic analysis shows that N. sphaerocarpa is located at a basal position of the genus Nitraria.
Nitraria sphaerocarpa Maxim. (Nitrariaceae) is a deciduous and drought-tolerant shrub. It occurs in deserts, foothills, gravelly and sandy areas of north-western China. Its sterile branches have spiny at apex, leathery leaves, and white flowers. When the fruit is ripe, the exocarp becomes membranous and expand into a ball, a unique morphological feature of N. sphaerocarpa that differs from the other four species of Nitraria in China (Liu and Zhou Citation2008). Nitraria sphaerocarpa is resistant to drought and sand. It is commonly used to stabilize sand dunes in north-western China. There are around nine species in the genus Nitraria from Western Sahara to Central Asia and Australia. We acquired the complete chloroplast (cp) genome by genome skimming approach, which provides resources for studying the phylogenetic evolution and speciation of this taxon.
In this study, the sample of N. sphaerocarpa was collected from Heshuo, Xinjiang Uygur Autonomous Region (42°13′33.94″N, 87°20′49.59″E). The voucher specimen was deposited in the Herbarium of the Xinjiang Institute of Ecology and Geography, Chinese Academy of Sciences (XJBI, Ying Feng, [email protected]), with a collection number of NL-1-6. Total genomic DNA was extracted from approximately 100 mg of silica-dried leaves material with a modified CTAB method (Doyle Citation1987). DNA extracts were fragmented for 300 bp short-insert library construction and sequenced 2 × 150 bp paired-end reads on an Illumina HiSeq X-Ten instrument at Beijing Genomics Institute (Shenzhen, China). The filtering of raw reads uses Trimmomatic 0.35 (Bolger et al. Citation2014) to remove adapters and low-quality bases. Then, about 3.0 GB clean reads were assembled using GetOrganelle (Jin et al. Citation2018). The finished cp genomes were annotated with GeSeq (Tillich et al. Citation2017) and adjusted manually using Geneious v 11.0.2 (Ripma et al. Citation2014), with the reference genome of N. tangutorum (MH457633). Finally, the annotated cp genomes were submitted to GenBank (Accession number: MW820161). MISA-web v2.1 (Beier et al. Citation2017) was applied to identify simple sequence repeats sequences (SSR).
The whole cp genome of N. sphaerocarpa is 159,369 bp in size (37.3% GC contents), with a typical quadripartite genome organization, including a large-single copy (LSC, 87,854 bp, 35.2% GC contents) region, a small-single copy (SSC, 18,383 bp, 31.4% GC contents) region, and a pair of two inverted repeats (IR, 26,566 bp, 42.7% GC contents) regions. A total of 80 SSRs were detected, 75 of which were mono-nucleotide (A/C/T, 93.75%), four were di-nucleotides (AT/TA, 5.0%), one was tri-nucleotides (AAT, 1.25%), respectively. The cp genome encoded 114 unigenes, including 79 protein-coding genes, 30 tRNA genes, four rRNA genes, and one pseudogene (infA). Among these genes, 14 genes contained one intron and three contained two introns (rps12, clpP, and ycf3).
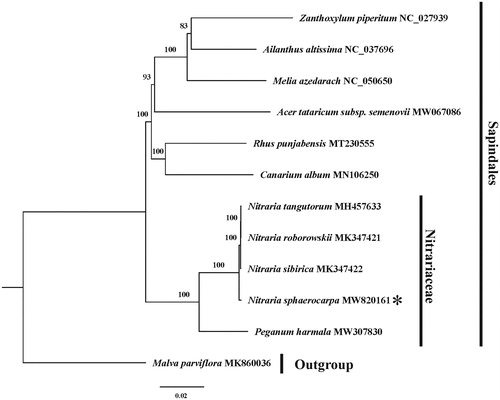
To investigate the phylogenetic position of N. sphaerocarpa, we performed a phylogenetic analysis based on complete cp genomes of five Nitrariaceae species and six species representing other 6 families within Sapindales. One species of Malvales was chosen as an outgroup. The DNA sequences for these 12 complete chloroplast genomes (after removing one IR) were aligned using the default option implemented in MAFFT version 7 (Katoh and Standley Citation2013). The most appropriate model (GTR + I + G) was determined by jModeltest v 2.1.10 (Darriba et al. Citation2012), and then Maximum likelihood (ML) tree was generated in RAxML 8.2.10 (Stamatakis Citation2014) with 1000 replicates (). The phylogenetic tree showed that Nitrariaceae is in a basal position among the seven families of Sapindales. Consistent with Zhang et al. (Citation2015), N. sphaerocarpa was located at a basal position of this genus. Besides, to find hyper-variable regions among four species of Nitraria, the sliding window analysis was performed by DnaSP v6 (Rozas et al. Citation2017), with a 1000 bp window and a 600 bp step size. Four hyper-variable regions (Pi > 0.004) in these genomes were identified, two of which are intergenic regions (trnHGUG-psbA and rpoB-trnCGCA, located in the LSC region), and two are protein-coding regions (ndhF and ycf1, located in the SSC and IR region, respectively). These hyper-variable regions can be great potential as taxon-specific barcodes for species identification within Nitraria genus.
Figure 1. The maximum-likelihood (ML) phylogenetic tree of 12 species (11 of Sapindales and one from Malvales was chosen as outgroup) based on complete chloroplast genomes (only one IR region). The number above branches are bootstrap support values.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The data that support the findings of this study are openly available in GenBank at https://www.ncbi.nlm.nih.gov/genbank/, accession number: MW820161. The raw sequencing data is available under GenBank Bioproject PRJNA728758.
Additional information
Funding
References
- Beier S, Thiel T, Munch T, Scholz U, Mascher M. 2017. MISA-web: a web server for microsatellite prediction. Bioinformatics. 33(16):2583–2585.
- Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 30(15):2114–2120.
- Darriba D, Taboada GL, Doallo R, Posada D. 2012. jModelTest 2: more models, new heuristics and parallel computing. Nat Meth. 9(8):772–772.
- Doyle J. 1987. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull. 19:11–15.
- Jin JJ, Yu WB, Yang JB, Song Y, Yi TS, Li DZ. 2018. GetOrganelle: a simple and fast pipeline for de novo assembly of a complete circular chloroplast genome using genome skimming data. bioRxiv. 4:256479.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30(4):772–780.
- Liu YX, Zhou LH. 2008. Nitrariaceae. In: Wu ZY, Raven PH, Hong D, editors. Flora of China, vol. 11. Beijing: Science Press, Missouri Botanical Garden Press.
- Ripma LA, Simpson MG, Hasenstab-Lehman K. 2014. Geneious! Simplified genome skimming methods for phylogenetic systematic studies: a case study in Oreocarya (Boraginaceae). Appl Plant Sci. 2(12):1400062.
- Rozas J, Ferrer-Mata A, Sã Nchez-DelBarrio JC, Guirao-Rico S, Librado P, Ramos-Onsins SE, N-Ga S. 2017. DnaSP 6: DNA sequence polymorphism analysis of large datasets. Mol Biol Evol. 34(12):3299–3302.
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30(9):1312–1313.
- Tillich M, Lehwark P, Pellizzer T, Ulbricht-Jones ES, Fischer A, Bock R, Greiner S. 2017. GeSeq – versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 45(W1):W6–W11.
- Zhang ML, Temirbayeva K, Sanderson SC, Chen X. 2015. Young dispersal of xerophil Nitraria lineages in intercontinental disjunctions of the Old World. Sci Rep. 5:13840.