
Abstract
In this paper, the complete mitochondrial genome of Ternate False Fusus Brunneifusus ternatanus Gmelin, 1791 was determined and characterized for the first time from the South China Sea. Our results showed that the length of the whole mitogenome of B. ternatanus was 16,254 bp and the mitogenome consisted of 22 tRNA genes, 13 protein-coding genes (PCGs), and 2 rRNA genes. Furthermore, the nucleotide composition of this mitogenome is significantly biased with G + C contents of 31.85% (the base content was 30.38% A, 16.17% G, 37.77% T, and 15.68% C). All PCGs shared ATG as the initiation codon, and stop codon of TAA or TAG, with the exception of COX2 which ended with a single T. The phylogenetic tree showed that B. ternatanus was first clustered with Hemifusus tuba, and from a single distinct cluster. The new complete mitochondrial genome provides new insight into the phylogenetic of Melongenidae and its evolution.
Brunneifusus ternatanus Gmelin, 1791, also known as Ternate False Fusus, is a species of sea snails, marine gastropod mollusks in the family Melongenidae. The scientific name, B. ternatanus, has only recently been accepted (previously named Hemifusus ternatanus) as a result of emerging research (Dekkers Citation2018). The characteristic features of B. ternatanus are similar to Hemifusus tuba, but it is longer and slenderer, usually smooth and without angular whorls, with a longer siphonal canal which is about 2/3 portion of the shell. Brunneifusus ternatanus is widely distributed in Western Pacific, especially in Japan (Phillips and Depledge Citation1986), Singapore (Chan Citation2009), Malaysia (Vermeij and Raven Citation2009), and China. It is found in the subtidal deep-water zone at a depth of 10–70 m. Because they are sold at a high price in markets, it has caused their overfishing. The taxonomy and phylogeny of the Melongenidae even Buccinoidea have been debated due to the lack of reliable morphological taxonomic characters (Kantor Citation2003). Here, we report the new complete mitochondrial genome of Melongenidae, which will provide a better insight into phylogenetic assessment and taxonomic classification.
The specimens of B. ternatanus were collected from Huanqiu fishing port of Wenchang, China (N19°33′51.12″, E110°49′27.98″), and deposited in the Hainan Marine Science and Technology Museum (Hongtao Liu, [email protected]) under the voucher number S20201203BT in Qionghai research base of Hainan Academy of Ocean and Fisheries Sciences. The de novo DNA libraries of B. ternatanus had an average length of 350 bp were constructed using the NexteraXT DNA Library Preparation Kit, and sequencing was performed on the Illumina Novaseq platform (Total Genomics Solution Limited, SZHT) generating reads of 150 bp in average length. The complete mitochondrial genome of B. ternatanus was assembled with 4.35 G clean reads using the de novo assembler SPAdes 3.11.0 (Bankevich et al. Citation2012) and was annotated using the MITOS (http://mitos.bioinf.uni-leipzig.de/index.py). Based on 13 protein-coding genes (PCGs) encoded by 45 species mitogenomes available in the GenBank, the phylogenetic analysis was carried out using IQ-TREE v1.6.12 (Nguyen et al. Citation2015) with the maximum-likelihood (ML) method used to investigate the evolution position of B. ternatanus. The bootstrap replicate was set as 1000, the best-fit model was chosen mtMet + F+R5 according to Bayesian information criterion (BIC).
The whole mitogenome of B. ternatanus is 16,254 bp in size (GenBank Accession No. MW548267). The overall base content was 30.38% A, 16.17% G, 37.77% T, and 15.68% C respectively. The 68.15% of (A + T) showed a high AT bias. The sequences of the B. ternatanus mitogenome consist of 22 tRNA genes, two rRNA genes, and 13 protein-coding genes (PCGs). Eight tRNA genes were located on the light strand, the others were encoded by the heavy strand.
The 22 tRNA genes in the mitogenome of B. ternatanus vary in length from 62 bp to 71 bp. There were two type copies of tRNA-Leu and tRNA-Ser, respectively, in the mitogenome. The 12S rRNA was 889 bp long and located between tRNA-Val and tRNA-Glu. The 16S rRNA was 1386 bp long and located between tRNA-Val and tRNA-Leu. All 13 PCGs shared a common initiation codon ATG. Simultaneously, 12 PCGs were terminated with the common stop codon TAA or TAG, except COX2 ended with a single T. There are 2 overlapping regions of 1–7 bp in length, and the longest overlapping region is located between ND4 and ND4L. The mitochondrial genome has 25 intergenic sequences varying from 1 to 935 bp in length. The largest intergenic sequence is located between tRNA-Phe and COX3 (Table S1).
The phylogenetic tree () showed that B. ternatanus formed a single distinct clade by clustering with Hemifusus tuba, and further clarified the phylogenetic relationships of the species of Melongenidae in the order Neogastropoda. Our phylogenetic result of B. ternatanus is consistent with the previous study using nuclear gene (18S rRNA and segment histone H3) and mitochondrial gene (COI, 12S rRNA, and 16S rRNA) (Zou et al. Citation2011). Taken together, the whole mitochondrial genome of B. ternatanus, characterized here, serves to clarify the phylogenetic relationships and comparative mitogenome studies of Melongenidae species in the Neogastropoda.
Figure 1. The maximum-likelihood tree of B. ternatanus and 45 other species based on 13 PCGs.
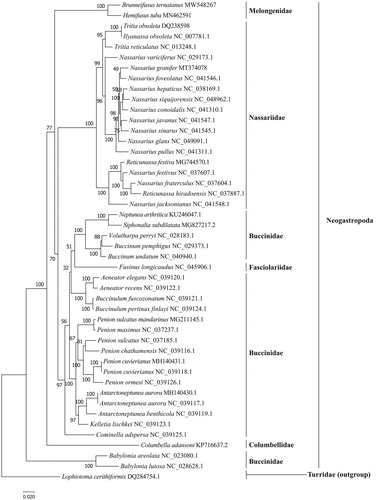
The GenBank accession number for each species is indicated after the scientific name. The ML tree was carried out using IQ-TREE v1.6.12 with 1000 bootstrap replicates, the best-fit model is mtMet + F+R5. The bootstrap values were labeled at each branch node. Lophioyoma cerithiformis was used as an outgroup.
Disclosure statement
No potential conflict of interest was reported by the authors.
Data availability statement
The genome sequence data that support the findings of this study are openly available in GenBank of NCBI at https://www.ncbi.nlm.nih.gov/ under the accession no. MW548267. The associated BioProject, SRA, and Bio-Sample numbers are PRJNA698244, SRR13589096, and SAMN17709251 respectively.
Additional information
Funding
References
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, et al. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol . 19(5):455–477.
- Chan S. 2009. The Melongenidae (Mollusca: Gastropoda) of Singapore. Nat Singapore. 2:63–67.
- Dekkers A. 2018. Two new genera in the family Melongenidae from the Indo-Pacific and comments on the identity of Hemifusus zhangyii Kosuge, 2008 and Pyrula elongata Lamarck, 1822 (Gastropoda, Neogastropoda: Buccinoidea). Gloria Maris. 57(2):40–50.
- Kantor YI. 2003. Comparative anatomy of the stomach of Buccinoidea (Neogastropoda). J Mollusc Stud. 69(3):203–220.
- Nguyen L-T, Schmidt HA, Von Haeseler A, Minh BQ. 2015. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 32(1):268–274.
- Phillips D, Depledge M. 1986. Distribution of inorganic and total arsenic in tissues of the marine gastropod Hemifusus ternatanus. Mar Ecol Prog Ser. 34:261–266.
- Vermeij GJ, Raven H. 2009. Southeast Asia as the birthplace of unusual traits: the Melongenidae (Gastropoda) of northwest Borneo. Contrib Zool. 78(3):113–127.
- Zou S, Li Q, Kong L. 2011. Additional gene data and increased sampling give new insights into the phylogenetic relationships of Neogastropoda, within the caenogastropod phylogenetic framework. Mol Phylogenet Evol. 61(2):425–435.