
Abstract
The complete mitogenome of Ips calligraphus was sequenced, the length was 19,144 bp which consists of 13 protein-coding genes, 22 transfer RNA genes, two ribosomal RNA genes, and a major non-coding AT-rich region (GenBank accession no. MW589547). All of 13 protein-coding genes (PCGs) started with ATN. 12 PCGs used the typical stop codon ‘TAA,’ while ATP8 terminated with stop codon ‘TAG.’ Phylogenetic analyses were performed using mitochondrial PCGs for the I. calligraphus and other 18 species within the Scolytinae. The I. calligraphus was clustered together with the other two Ips species in tribe Ipini which were closely related to Xyleborini and Dryocoetini.
Ips calligraphus (Germar 1824) is mainly a secondary pest of conifer forests and feeds mostly on Pinus spp. Ips calligraphus was native in North and Central America and the Caribbean Islands (Wood and Bright Citation1992). It was introduced to Benguet, Philippines in 1987 (Zamora Citation1987) and found in Guangdong, China recently.
Adult individuals of I. calligraphus were collected from Fenghuang mountain in Zhuhai Guangdong, China (22.29 N 113.52 E) on 31 October 2019. A specimen was deposited at −20 °C in the plant quarantine laboratory of the Technical Center of Gongbei Customs District, P. R. China (http://gongbei.customs.gov.cn/, Wei Lin, [email protected]) under the voucher number: IC11.
The complete mitochondrial genome of Ips calligraphus (GenBank: MW589547) was sequenced using Illumina Novaseq 6000 platform and de novo assembly was conducted by MitoZ (Meng et al. Citation2019). The I. calligraphus mitochondrial genome is a closed circular molecule of 19,144 bp in length. The genome contains 13 Protein-coding genes (PCGs), 22 transfer RNA genes (tRNAs), two ribosomal RNA genes (rrnL and rrnS), and a non-coding AT-rich region. The complete mitochondrial nucleotide composition was biased to AT 75.7%, and the total base composition was 39.9% A, 35.8% T, 10.1% G, 14.2% C.
The total length of protein-coding genes was 10,716 bp. All PCGs start with the conventional initiation codons (ATN). Twelve PCGs shared the same complete stop codon (TAA), while the ATP8 gene used TAG as the stop codon. Individual tRNAs of I. calligraphus ranged from 63 bp (trnC) to 72 bp (trnE) in length. The large ribosomal gene (rrnL) was 1367 bp in length, which is located between trnL1 and trnV. The small ribosomal gene (rrnS) was 819 bp in length and positioned between trnV and the AT-rich region. The non-coding AT-rich region located in rrnS and trnI corresponding to the control region, with A + T contents, are 79.9%. Furthermore, there was an 1158 bp ‘supernumerary’ non-coding region between trnI and trnQ like some other species in Curculionidae.
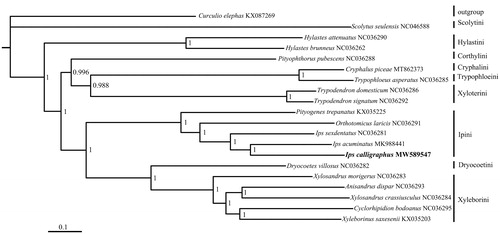
Phylogenetic analysis of I. calligraphus with 18 other Scolytinae species was conducted based on nucleotide sequence data of 13 PCGs (). The analysis was performed with Bayesian inference under GTR + F + I + G4 model in Phylosuite (Zhang et al. Citation2020). The phylogenetic tree showed that the relationship within Scolytinae was similar to the previous study (Vega and Hofstetter Citation2015). The I. calligraphus was clustered together with the other two Ips DeGeer, 1775 species in tribe Ipini which were closely related to Xyleborini and Dryocoetini.
Figure 1. Phylogenetic tree showing the relationship between Ips calligraphus and other Scolytinae species based on Bayesian methods. Numbers on branches are Bayesian posterior probabilities.
Acknowledgments
The authors thank You Li and Andrew J. Johnson (University of Florida) for assistance with beetle identification.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The genome sequence data that support the findings of this study are openly available in GenBank of NCBI at (https://www.ncbi.nlm.nih.gov/) under the accession no. MW589547. The associated BioProject, SRA, and Bio-Sample numbers are PRJNA743429, SRR15093617, and SAMN20035122 respectively.
Additional information
Funding
References
- Meng G, Li Y, Yang C, Liu S. 2019. MitoZ: a toolkit for animal mitochondrial genome assembly, annotation and visualization. Nucleic Acids Res. 47(11):e63.
- Vega FE, Hofstetter RW. 2015. Biology, systematics, and evolution of Ips. In Bark beetles: biology and ecology of native and invasive species. San Diego (CA): Elsevier; p. 341–370.
- Wood SL, Bright DE. 1992. A catalog of Scolytidae and Platypodidae (Coleoptera), Part 2. Taxonomic Index. Great Basin Naturalist Memoirs. Provo (UT): Brigham Young University; p. 484–538.
- Zamora RA. 1987. Life history and phenology of Ips calligraphus Germar in Benguet province [Philippines]. Phil Technol J. 12:43–60.
- Zhang D, Gao F, Jakovlić I, Zou H, Zhang J, Li WX, Wang GT. 2020. PhyloSuite: an integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol Ecol Resour. 20(1):348–355.