
Abstract
The complete chloroplast genome of Salix cupularis Rehder was assembled and subjected to phylogenetic analysis. The chloroplast genome of S. cupularis was 155,518 bp in length, containing a large single-copy region (84,373 bp), a small single-copy region (16,226 bp), and two inverted repeat regions (27,458 bp). The overall GC content of S. cupularis chloroplast genome was 36.70%. The chloroplast genome of S. cupularis contained 127 unique genes, including 82 protein-coding genes, 37 tRNA genes, and eight rRNA genes. Phylogenetic analysis showed that S. cupularis was most related to Salix magnifica.
Salix cupularis Rehder is a typical alpine shrub, belongs to Salicaceae, and is mainly distributed in alpine hillslope of 2540–4000 m in Sichuan, Gansu and Qinghai Province, China. As the shrub can grow in extremely harsh alpine semiarid environments, playing an important role in fixing sand, resisting wind erosion, increasing plant diversity and preserving water, S. cupularis has been being used as a sand binder in alpine hillslope (Hu et al. Citation2018). To make better use of S. cupularis, the complete chloroplast genome of S. cupularis was assembled and subjected to phylogenetic analysis.
Fresh leaves of S. cupularis were sampled from Kangding City, northwest Sichuan Province, China (N30°6′44.68″, E101°45′25.29″). Total genomic DNA was extracted according to the mCTAB protocol (Li et al. Citation2013). The specimen (Voucher No. SCRA-202007091) and its genome DNA (Accession number: No. SCRA-202007091G) were deposited in the Laboratory of Forest Pathology, College of Forestry, Sichuan Agricultural University (Zhihang Zhuo and [email protected]). The genome sequencing was conducted using Illumina NovaSeq platform by Shanghai Personal Biotechnology Co. Ltd, China. In all, 3.3 G raw reads were obtained, and after the chloroplast genome of S. cupularis was assembled with SPAdes v3.9.0 software (Bankevich et al. Citation2012) and annotated with Plann (Huang and Cronk Citation1975). The complete chloroplast genome sequence of S. cupularis has been submitted to the GenBank (MW307821).
The chloroplast genome of S. cupularis was 155,518 bp in length, containing a large single-copy (LSC) region (84,373 bp), a small single-copy (SSC) region (16,226 bp), and two inverted repeat (IR) regions (27,458 bp). The overall GC content of chloroplast genome was 36.70%, and in the SSC, LSC, and IR regions were 31.00%, 34.40%, and 41.90%, respectively. The chloroplast genome contained 127 complete genes, including eight rRNA genes, 37 tRNA genes, and 82 protein-coding genes.
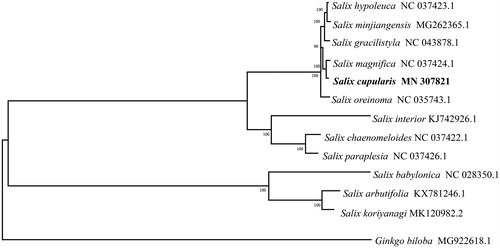
To evaluate the phylogenetic relationships of S. cupularis within Salicaceae, the chloroplast genome sequence of S. cupularis and those of 11 other Salicaceae species were aligned by MAFFT v7 (Katoh and Standley Citation2013), and the phylogenetic tree was constructed with MEGA X using neighbour-joining analysis with 1000 bootstrap replicates (Kumar et al. Citation2018). The phylogenetic tree revealed that S. cupularis was mostly related to S. magnifica ().
Figure 1. The phylogenetic relationship of 12 species within the Salicaceae species based on neighbour-joining analysis of complete chloroplast genomes. Ginkgo biloba was served as the out-group.
Date availability statement
The genome sequence data that support the findings of this study are openly available in GenBank of NCBI at (https://www.ncbi.nlm.nih.gov/) under the accession number MW307821 (https://www.ncbi.nlm.nih.gov/nuccore/MW307821). The associated BioProject, SRA, and Bio-Sample numbers are PRJNA688100, SRS7935879, and SAMN17168987 respectively.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, et al. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 19(5):455–477.
- Hu YF, Shu XY, He J, Zhang YL, Xiao HH, Tang XY, Gu XF, Lan T, Xia JG, Ling J, et al. 2018. Storage of C, N and P affected by afforestation with Salix cupularis in an alpine semi-arid desert ecosystem. Land Degrad Dev. 29(1):188–198.
- Huang DI, Cronk QCB. 1975. Plann: a command-line application for annotating plastome sequences. Biochem Pharmacol. 24(17):1639–1641.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30(4):772–780.
- Kumar S, Stecher G, Li M, Knyaz C, Tamura K. 2018. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 35(6):1547–1549.
- Li J, Wang S, Jing Y, Wang L, Zhou S. 2013. A modified CTAB protocol for plant DNA extraction. Chin Bull Bot. 48(1):72–78.