
Abstract
Callitriche palustris L. is an annual aquatic or marsh plant, wide spread in temperate regions throughout the world. In present study, we sequenced, assembled and annotated the complete chloroplast (cp) genome of C. palustris. The length of C. palustris complete cp genome was 150,138 bp, with a typical quadripartite structure comprising a pair of inverted repeat regions (IRs; 25,667 bp), a large single copy region (LSC; 81,432 bp) and a small single copy region (SSC; 17,372 bp). The whole cp genome contained 134 genes, including 89 protein-coding genes (PCGs), 37 transfer RNA (tRNA) genes, and eight ribosomal RNA (rRNA) genes. The maximum likelihood (ML) phylogenetic analysis indicated that C. palustris was a member of Plantaginaceae, but the relationships between subfamilies and tribes need more samplings. This cp genome would provide a valuable genetic resource for C. palustris’ phylogenetic study.
Callitriche palustris L. is an annual aquatic or marsh plant, with very small flowers in the axillary. It is wide spread in temperate regions throughout the world, in the altitude from sea level to 5000 m (Min and Lansdown Citation2008). Pollination of the aquatic Callitriche may be by wind (above the lake surface) or by water, or there may be self-pollination (Martinsson Citation1993; Osborn et al. Citation2001). Callitriche was the only representative in Callitrichaceae, but now is placed in the family Plantaginaceae based on three plastid genes (rbcL, ndhF, and rps2) (Osborn et al. Citation2001).
The voucher specimen of C. palustris used in this study were collected from Bashui, Huangzhou, Hubei, China (115°22′03.31″E, 31°11′19.92″N, 20 m, in the pool). The collected specimen was deposited in Herbarium of Huanggang Normal University (HGTC, former Herbarium of Biology Department of Huanggang Teachers College, Mr. Jun Fu, [email protected]) under voucher number 2018-12-1 (collected by Dong Hongjin et al.). Genomic DNA was extracted from fresh leaves of a seedling according to a modified CTAB method (Doyle and Doyle Citation1987). Total genome DNA of C. palustris was sequenced by Illumina Hiseq 2500 Sequencing System (Illumina, Hayward, CA) with 150 bp paired-end. The reads were assembled through the GetOrganelle software (Jin et al. Citation2020). The complete cp genome of C. palustris was annotated with software PGA (Qu et al. Citation2019) and Geneious ver. 10.1 (http://www.geneious.com (Matthew et al. Citation2012) and then submitted to GenBank (accession no. MW774642). The genome annotation was performed by aligning with the cp genomes of relatively related species.
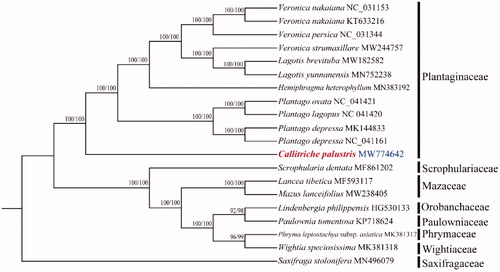
The size of cp genome of C. palustris is 150,138 bp, including a large single-copy (LSC) region of 81,432 bp and a small single-copy (SSC) region of 17,372 bp separated by a pair identical inverted repeat regions (IRs) of 25,667 bp each. A total of 134 genes were successfully annotated containing 89 protein-coding genes, 37 tRNA genes, and eight rRNA genes. GC content of the whole genome, IRs, LSC and SSC regions are 37.8%, 43.1%, 37.8%, and 31.0%, respectively. GC content of IRs region is the highest. Twenty-one genes contain one intron, while two genes have two introns. The complete cp genome sequence of C. palustris and other species from Plantaginaceae and close relatives were used to construct phylogenetic tree (). The sequences were initially aligned using MAFFT (Kazutaka and Standley Citation2013) and then visualized and manually adjusted using BioEdit (Hall Citation1999). Take the plastome of Saxifraga stolonifera (GenBank: MN496079) as an out-group, a maximum likelihood analysis was performed with RAxML version 8 program (Alexandros Citation2014) using 1000 bootstrap. IQ-tree was also used to construct ML tree with fast mode (Nguyen et al. Citation2015). The result supports that C. palustris was a member of Plantaginaceae, consistent with the previous studies (Philbrick and Les Citation2000; Olmstead et al. Citation2001). The relationships between subfamilies and tribes need more samplings.
Figure 1. Maximum likelihood phylogenetic tree for Callitriche palustris based on complete cp genomes. The number on each node indicates bootstrap support value generated by RaxML/IQ-tree.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The data that support the findings of this study are available in GenBank of NCBI at https://www.ncbi.nlm.nih.gov, accession number MW774642. The assembled individual was linked with no. SAMN18324991 and Project ID: PRJNA715046. Raw sequencing reads used in this study were deposited in the GenBank database of Sequence Read Archive with no. SRR14844933.
Additional information
Funding
References
- Alexandros S. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30(9):1312–1313.
- Doyle JJ, Doyle JL. 1987. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochemical Bulletin. 19(1):11–15.
- Hall TA. 1999. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser. 41:95–98.
- Jin JJ, Yu WB, Yang JB, Song Y, dePamphilis C, Yi TS, Li DZ. 2020. GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 21(1):241.
- Kazutaka K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30(4):772–780.
- Martinsson K. 1993. The pollen of Swedish Callitriche (Callitrichaceae) – trends towards submergence. Grana. 32(4–5):198–209.
- Matthew K, Richard M, Amy W, Steven SH, Matthew C, Shane S, Simon B, Cooper A, Sidney M, Chris D. 2012. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 28(12):1647–1649.
- Min T, Lansdown R. 2008. Callitrichaceae. In: Wu ZY, Raven PH, editors. Flora of China. Beijing: Science Press; p. 318–320.
- Nguyen LT, Schmidt HA, Arndt VH, Bui Quang M. 2015. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 32(1):268–274.
- Olmstead R, dePamphilis C, Wolfe A, Young N, Elisons W, Reeves P. 2001. Disintegration of the Scrophulariaceae. Am J Bot. 88(2):348–361.
- Osborn J, El-Ghazaly G, Cooper R. 2001. Development of the exineless pollen wall in Callitriche truncata (Callitrichaceae) and the evolution of underwater pollination. Plant Systematics and Evolution. 228(1-2):81–87.
- Philbrick T, Les D. 2000. Phylogenetic studies in Callitriche: implications for interpretation of ecological, karyological and pollination system evolution. Aquat Bot. 68(2):123–141.
- Qu XJ, Moore MJ, Li DZ, Yi TS. 2019. PGA: a software package for rapid, accurate, and flexible batch annotation of plastomes. Plant Methods. 15(1):1–12.