
Abstract
Rubus rufus Focke is a deciduous shrub species native to subtropical China and is valuable resources for the Raspberry cultivation by virtue of prolific fruits and stout resistances to hash stress. In this study, we assembled and characterized the complete chloroplast genome of R. rufus as resources for the future study. The chloroplast genome was 156,232 bp in length, composing of one large single copy (LSC; 85,840 bp) and one small single copy (SSC; 18,850 bp), separated by two inverted repeats (IRs; 25,771 bp). A total of 130 genes were predicted, including eight rRNAs, 36 tRNAs, and 86 protein-coding genes. The phylogenetic analysis showed a close relationship between R. rufus and Rubus cochinchinensis in the tribe Rubeae.
Rubus rufus Focke is a climbing shrub native to subtropical China. This species shows great morphological variations, and three varieties have been recognized so far (Lu and Boufford Citation2003). With prolific fruits and stout resistances to diverse hash stress (Personal observation), R. rufus holds great potential for Raspberry cultivation in the future. However, the paucity of genetic resources has hindered the studies on phylogeny, diversity and speciation for this species. Here, we assembled and characterized the complete chloroplast genome sequence of R. rufus as a resource for future studies on this species.
We sampled leaves of R. rufus var. rufus from a forest understory at the Dawei Mountain in Yunnan Province (22°75' N, 103°45' E, alt. 750 m). A voucher specimen was also deposited at the Sun Yat-sen University Herbarium (SYS) with accession number RCOC-18274-XN05 (Weicheng Huang, [email protected]). Total genomic DNA was extracted from silica gel–dried leaves using a modified CTAB protocol (Doyle Citation1991). Using the qualified DNA, a library with insertion size of 500 bp was constructed and sequenced with paired-end (150 bp) read on an Illumina Hiseq 3000 platform (San Diego, CA, USA) at Jierui Bio-Technique Co. Ltd (Beijing, China). Prior to the chloroplast genome assembly, all raw reads were filtered and trimmed with default parameters using the package fastp v0.20.1 (Chen et al. Citation2018). Then, the clean reads were mapped to the published chloroplast genome of Rubus leucanthus (MK105853.1) using BWA v0.7.17 (Li and Durbin Citation2009), and the obtained reads were fed to package Unicycler v0.4.8 (Wick et al. Citation2017) for chloroplast genome assembly. Next, the obtained contigs were oriented by aligning to R. leucanthus chloroplast genome using Mummer 3.2 (Kurtz et al. Citation2004). Finally, the genome assemblies were annotated using package Geseq (Tillich et al. Citation2017) and corrected manually using Geneious v.9.0.2 (Kearse et al. Citation2012). The complete chloroplast genome sequence of R. rufus was submitted to GenBank with the accession number of MW930397.
The complete chloroplast genome of R. rufus was 156,232 bp in length, with an 85,840 bp large single copy (LSC) and a 18,850 bp small single copy separated by two inverted repeats (25,771 bp). The chloroplast genome circle is joined end to end in the order of 'LSC-IRA-SSC-IRB'. The overall GC content is 37.18%, A total of 130 genes were predicted, consist of 86 protein-coding genes, 36 tRNA genes and eight rRNA genes.
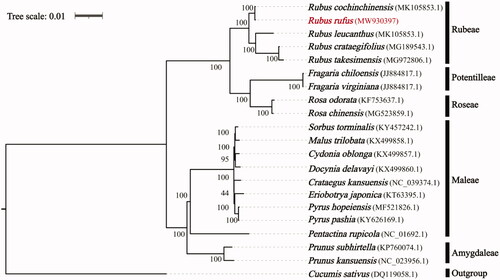
For phylogenetic analysis, the chloroplast genomes of R. fufus, 19 other Roseaceae species, and one outgroup species Cucumis sativus (DQ119058.1) were downloaded from GenBank. All the sequences were aligned using MAFFT v.7.313 (Katoh and Standley Citation2013) and a maximum-likelihood phylogenetic analysis was implemented using RAxML v.8.2.11 (Stamatakis Citation2014) (). The phylogeny analysis demonstrated that R. rufus was placed in the Rubeae tribe with close relationship with Rubus cochinchinensis, and Rubeae tribe was monophyly with high support for among the five tribes used in this study.
Figure 1. Maximum-likelihood tree of Rosaceae base on complete chloroplast genomes, with Cucumis sativus as outgroup. The Rubus rufus is marked in red and Bootstrap support values (based on 1000 replicates) are shown next to the nodes.
Acknowledgement
The authors declare that there is no conflict of interest regarding the publication of this article. The authors alone are responsible for the content and writing of the paper.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The genome sequence data that support the findings of this study are openly available in GenBank of NCBI at https://www.ncbi.nlm.nih.gov/ under the accession no. MW930397. The associated BioProject, SRA, and Bio-Sample numbers are PRJNA739406, SRR14883822, and SAMN19789077, respectively.
Additional information
Funding
References
- Chen S, Zhou Y, Chen Y, Gu J. 2018. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 34(17):i884–i890.
- Doyle JJ. 1991. DNA protocols for plants—CTAB total DNA isolation. In: Hewitt GM, Johnston A (eds) Molecular techniques in taxonomy. Berlin: Springer. p. 283–293.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30(4):772–780.
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, et al. 2012. Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 28(12):1647–1649.
- Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, Antonescu C, Salzberg SL. 2004. Versatile and open software for comparing large genomes. Genome Biol. 5(2):R12.
- Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 25(14):1754–1760.
- Lu LD, Boufford DE. 2003. Rubus Linnaeus. In: Wu ZY. & Rave PH. (eds.) Flora of China, vols. 9. St. Louis: Science Press, Beijing & Missouri Botanical Garden Press. p. 46–434.
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30(9):1312–1313.
- Tillich M, Lehwark P, Pellizzer T, Ulbricht-Jones ES, Fischer A, Bock R, Greiner S. 2017. GeSeq - versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 45(W1):W6–W11.
- Wick RR, Judd LM, Gorrie CL, Holt KE. 2017. Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput Biol. 13(6):e1005595.