
Abstract
Lessertia frutescens (L.) Goldblatt & J. C. Manning 2000 is an endemic species of Southern Africa with high medicinal and economic values. To facilitate exploration of its genetic resource, a complete chloroplast genome was determined using Illumina pair-end sequencing technology. The complete circular genome is 122,700 bp in length with overall 34.2% GC contents. It encodes a total of 110 genes, including 76 protein-coding genes, 30 tRNA, and four rRNA genes. The maximum-likelihood (ML) phylogenetic tree indicated that L. frutescens nested within the Papilionoideae and had a close relationship with Astragalus nakaianus and A. mongholicus. The newly sequenced complete chloroplast genome will help understanding the plastome evolution, genetic diversity and contribute to the genetic conservation of the natural population of L. frutescens.
Lessertia frutescens (L.) Goldblatt & J. C. Manning 2000, known as “cancer bush,” is an indigenous Southern Africa perennial shrub with attractive flowers (van Wyk and Albrecht Citation2008), belonging to the inverted repeat lacking clade (IRLC) of the Leguminosae (Legume Phylogeny Working Group [LPWG] Citation2017). It had been found to contain a large number of phytochemicals and several beneficial properties which used to treat a variety of ailments including stomach complaints, diabetes and uterine troubles. Notably, it had been used to treat and prevent cancer since 1895(van Wyk and Albrecht Citation2008). Traditionally, L. frutescens represents a variable species complex that is divided into three subspecies and several regional forms (van Wyk and Albrecht Citation2008). Therefore, L. frutescens requires more morphological and genetic evidence to distinguish the variable species complex and other sub-taxa. Chloroplast genome provides a powerful tool for reconstructing phylogenetic relationships and the development of molecular makers for the identification of plant species (Jansen et al. Citation2007; Huang et al. Citation2020). In the present study, the complete chloroplast genome of L. frutescens was studied which significantly contributed toward the phylogeny, genetics conservation and provide genetic resources for polymorphism investigations of this complex species.
The sample was collected from a cultivated individual in Kirstenbosch Botanical Garden, Cape Town, South Africa (33°57′41″S, 18°24′37″E). The voucher specimens were deposited in the herbarium of Kunming Institute of Botany, Chinese Academy of Sciences (http://www.kun.ac.cn/, Tao Deng, [email protected]) under the voucher number G15518. Total genomic DNA was isolated with a modified CTAB protocol (Doyle and Doyle Citation1987) and stored in the −80 degree refrigerator of Germplasm Bank of Wild Species, Kunming Institute of Botany, Chinese Academy of Sciences (http://www.genobank.org/, Rong Zhang, [email protected]). Part of DNA was sent to Beijing Genomics Institute, Shenzhen, China for constructing a paired-end (PE) library and sequencing using the Illumina HiSeq2000. The chloroplast genome was assembled and annotated following Zhang et al. (Citation2020). Arachis hypogaea L. (GenBank: NC026676) was selected as reference genome. Bandage Linux v.8.0 (Wick et al. Citation2015) was used to assemble the contigs and Bowtie2 (Langmead and Salzberg Citation2012) was used for mapping PE reads to the chloroplast genome. Finally, annotation was performed on GeSeq (Tillich et al. Citation2017), coupled with manual adjustment in Geneious v.9.1.4 (Kearse et al. Citation2012) and the chloroplast genome with accession number MF286764 was submitted to GenBank.
The complete chloroplast genome of L. frutescens was 122,700 bp in length which lacked the inverted repeat structure. A total of 110 genes were encoded, including 76 protein-coding genes, 30 tRNAs and four rRNAs. Similar to other IRLC chloroplast genomes, L. frutescens lost rpl22 and rps16 gene. Besides, its clpP gene lost the second intron, atpF and rps12 genes lost an intron. The overall GC content was 34.2% while the protein-coding regions (PCRs) were 65, with 670 bp in length and 53.5% of the total chloroplast genome. The percentage of PCRs was slightly higher than that of basal taxon Glycyrrhiza glabra L. (NC_024038, 66,600/127,943 bp = 52.1%) of the IRLC.
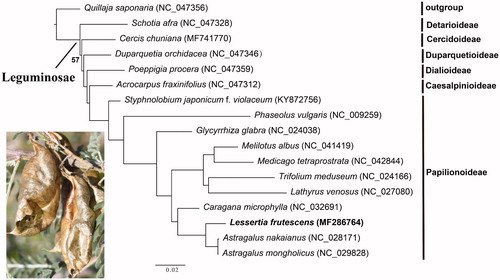
To compare the topology with limited samples at subfamily level to the result with extensive samples in Zhang et al. (Citation2020), a phylogenetic tree was constructed using RAxML v.8.2.12 (Stamatakis Citation2014) based on 16 plastomes representing the all six subfamilies of Leguminosae and closely related species of L. frutescens. Quillaja saponaria Molina was used as outgroup. The phylogenetic result () showed that L. frutescens nested within the subfamily Papilionoideae and formed a clade with Astragalus nakaianus and A. mongholicus with 100% bootstrap support. The subfamily Detarioideae was sister to other legumes with moderate support (57%), consistent with earlier studies based on coding genes from Zhang et al. (Citation2020). The reconstructed phylogeny also provided robust phylogenetic relationships among four other subfamilies of Leguminosae. The newly sequenced complete chloroplast genome will provide resources for phylogenetic reconstruction of the legume family and conservation of this important medicinal plant species.
Figure 1. Maximum likelihood (ML) phylogenetic tree based on 17 chloroplast genomes. ML bootstrap values <100% are shown. The position of the newly sequenced Lessertia frutescens is shown in bold. The photograph was taken during a field collection of L. frutescens.
Acknowledgments
We gratefully acknowledge the Plant Germplasm and Genomics Center, Kunming Institute of Botany, Chinese Academy of Sciences (KIB, CAS), for DNA extraction.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The genome sequence data of L. frutescens are openly available in GenBank of NCBI [https://www.ncbi.nlm.nih.gov] (https://www.ncbi.nlm.nih.gov/) under the accession no. MF286764, the associated BioProject, SRA, and Bio-Sample numbers are PRJNA739544, SRR14868908, and SAMN19791851 respectively.
Additional information
Funding
References
- Doyle JJ, Doyle JL. 1987. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull. 19:11–15.
- Huang R, Liang Q, Wang Y, Yang T-J, Zhang Y. 2020. The complete chloroplast genome of Epimedium pubescens Maxim. (Berberidaceae), a traditional Chinese medicine herb. Mitochondrial DNA B Resour. 5(3):2042–2044.
- Jansen RK, Cai Z, Raubeson LA, Daniell H, Depamphilis CW, Leebens-Mack J, Müller KF, Guisinger-Bellian M, Haberle RC, Hansen AK, et al. 2007. Analysis of 81 genes from 64 plastid genomes resolves relationships in angiosperms and identifies genome-scale evolutionary patterns. Proc Natl Acad Sci U S A. 104(49):19369–19374.
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, et al. 2012. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 28(12):1647–1649.
- Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods. 9(4):357–359.
- Legume Phylogeny Working Group [LPWG]. 2017. A new subfamily classification of the Leguminosae based on a taxonomically comprehensive phylogeny. Taxon. 66:44–77.
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30(9):1312–1313.
- Tillich M, Lehwark P, Pellizzer T, Ulbricht-Jones ES, Fischer A, Bock R, Greiner S. 2017. GeSeq - versatile and accurate annotation of organelle genomes . Nucleic Acids Res. 45(W1):W6–W11.
- van Wyk B, Albrecht C. 2008. A review of the taxonomy, ethnobotany, chemistry and pharmacology of Sutherlandia frutescens (Fabaceae). J Ethnopharmacol. 119(3):620–629.
- Wick RR, Schultz MB, Zobel J, Holt KE. 2015. Bandage: interactive visualization of de novo genome assemblies. Bioinformatics. 31(20):3350–3352.
- Zhang R, Wang Y-H, Jin J-J, Stull GW, Bruneau A, Cardoso D, De Queiroz LP, Moore MJ, Zhang S-D, Chen S-Y, et al. 2020. Exploration of plastid phylogenomic conflict yields new insights into the deep relationships of Leguminosae. Syst Biol. 69(4):613–622.