
Abstract
Salix chienii W.C. Cheng (Salicaceae) is a dioecious small tree with high economic and medicinal value. In this study, we reported the complete chloroplast DNA (cpDNA) of S. chienii by employing HiSeq sequencing and de novo assembly. The chloroplast genome of S. chienii is 155,672 bp in length, with a 36.60% GC content; it contains one large single copy (LSC), one small single copy (SSC) and two inverted repeat (IR) regions with lengths of 84,494, 16,330, and 27,424 bp, respectively. A total of 132 genes were predicted, including 85 protein-coding genes, 37 tRNA genes, 8 rRNA genes and 2 pseudogenes. A maximum-likelihood phylogenetic tree showed that S. chienii is closely related to S. wilsonii and S. triandra.
Salix chienii W.C. Cheng belongs to the genus Salix, family Salicaceae, and is a unique tree species in China (Wang and Fang Citation1984). As a dioecious species, S. chienii can be found as shrubs on both sides of streams at altitudes of 500 to 600 meters in several provinces, including Anhui, Zhejiang, Hunan, Hubei, Jiangsu and Jiangxi. This plant is medically important and can be used for the treatment of fever, sore throat and skin pruritus (Xiao et al. Citation2004). However, to date, research on S. chienii has been limited, and no molecular studies of the species have been conducted. Consequently, the breeding and application of S. chienii are restricted, and this plant has not been artificially cultivated.
To promote the protection of S. chienii germplasm resources, we reported the complete chloroplast genome sequence of S. chienii (GenBank accession number: MW969692) based on Illumina paired-end sequencing in this study. We also determined its phylogenetic relationship with other species of the Salicaceae family to better understand their interrelation.
Fresh, healthy leaves of S. chienii were collected from Mount Huangshan (Anhui, China; coordinates: 118°17′25.75″E, 30°11′22.68″N), and the voucher specimen was deposited in Room 60708, Biotechnology Building, Nanjing Forestry University, China (Xiaoping Li, [email protected], HSYYL2020001). Total genomic DNA was extracted from the fresh leaves using the DNeasy Plant Mini Kit (Qiagen, Valencia, CA, USA) and was further used to construct an Illumina paired-end (PE) genome library. Whole-genome sequencing was conducted with 150 bp paired-end reads on the Illumina NovaSeq 6000 Platform (Illumina, USA) (Genepioneer Biotechnologies Co. Ltd, Nanjing, Jiangsu, China). Approximately 13.4 G of raw reads were generated after sequencing. Low-quality reads were trimmed by using Fastp (Chen et al. Citation2018). The clean reads were assembled using the software NOVOPlast (Dierckxsens et al. Citation2017) and then annotated and corrected by using PGA (Plastid Genome Annotator) and Geneious Prime version 2021.1.1 (Qu et al. Citation2019; Kearse et al. Citation2012) through comparison with the complete chloroplast genome of its genetically close species S. babylonica (MF189169.1; Wang and Yang Citation2016).
The chloroplast genome of S. chienii was 155,672 bp in length and contained a small single-copy region (SSC; 16,330 bp), a large single-copy region (LSC; 84,494 bp) and a pair of inverted repeat regions (IR; 27,424 bp). The total GC content of the chloroplast genome was 36.6%, with different values for the SSC (31.0%), LSC (34.4%), and IR (41.7%) regions. A total of 132 genes were annotated from the chloroplast genome sequence, including 85 protein-coding genes, 37 transfer RNA (tRNA) genes, 8 ribosomal RAN (rRNA) genes and 2 pseudogenes. A total of 19 genes, including eight protein-coding genes, seven tRNAs and four rRNAs, were duplicated in the IR regions.
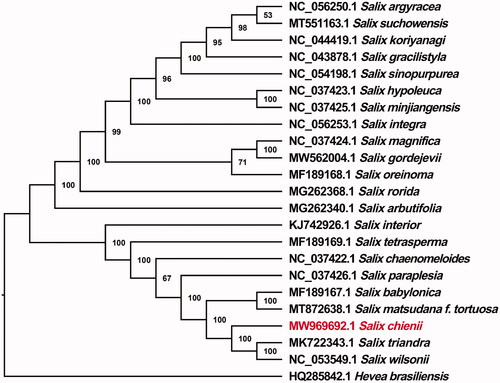
To explore the phylogenetic position of S. chienii among Salix, a phylogenetic analysis was performed by comparing the chloroplast genome sequences of S. chienii with 21 other Salicaceae species. All sequences were downloaded from the GenBank database and aligned by MAFFT v7 (Katoh and Standley Citation2013), and a phylogenetic tree was constructed by using IQ-TREE v2.1.3 (Nguyen et al. Citation2015) based on the maximum likelihood (ML) algorithm (with the best model of K3Pu + F+R8 and 1000 bootstrap replicates). In order to add a distinct root to the ML tree, Hevea brasiliensis (Tangphatsornruang et al. Citation2011) derived from Euphorbiaceae was set as an outgroup. The bootstrap support value (%) is shown next to the nodes. Our ML phylogenetic tree showed that S. chienii was closely related to the two congeners, S. wilsonii and S. triandra, with high bootstrap values (). The complete chloroplast genome of S. chienii presented here may contribute to the further study of genetic diversity, molecular identification and phylogenetic classification of the genus Salix (Salicaceae).
Figure 1. A maximum likelihood phylogenetic tree of S. chienii and 21 other Salicaceae species based on chloroplast genome sequences; Hevea brasiliensis was used as an outgroup. Bootstrap support values are indicated for each node.
Disclosure statement
No potential conflict of interest was reported by the authors.
Data availability statement
The sequence data that support the findings of this study are openly available in GenBack of NCBI https://www.ncbi.nlm.nih.gov/ under the accession no.MW969692. The associated BioProject, Bio-Sample and SRA numbers are PRJNA727759, SAMN19036906 and SRR14479090, respectively.
Additional information
Funding
References
- Chen S, Zhou Y, Chen Y, Jia G. 2018. Fastp: an ultra-fast all-in-one fastq preprocessor. Bioinformatics. 34(17):i884–i890.
- Dierckxsens N, Mardulyn P, Smits G. 2017. NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 45(4):e18.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30(4):772–780.
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, et al. 2012. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 28(12):1647–1649.
- Nguyen L-T, Schmidt HA, von Haeseler A, Minh BQ. 2015. Arndt von Haeseler, Bui Quang Minh, IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 32(1):268–274.
- Qu XJ, Moore MJ, Li DZ, Yi TS. 2019. PGA: a software package for rapid, accurate, and flexible batch annotation of plastomes. Plant Methods. 15:50.
- Tangphatsornruang S, Uthaipaisanwong P, Sangsrakru D, Chanprasert J, Yoocha T, Jomchai N, Tragoonrung S. 2011. Characterization of the complete chloroplast genome of Hevea brasiliensis reveals genome rearrangement, RNA editing sites and phylogenetic relationships. Gene. 475(2):104–112.
- Wang Chan & Fang Cheng-Fu, editors. 1984. Salicaceae. Fl. Reipubl. Popularis Sin. Beijing: China Science Press, Vol. 20. p. 1–403.
- Wang Y, Yang H. 2016. The complete chloroplast genome sequence of Salix babylonica. Mitochondrial DNA A DNA Mapp Seq Anal. 27(6):4683–4684.
- Xiao D, Liu C, Tan D, Han Y, Yin T, Lu Z. 2004. Pharmaceutical annals of Hunan. Vol 6. S. chienii. Hunan (China): Science and Technology Press; p. 304–305.