
Abstract
We report complete chloroplast genome (plastome) sequences of Stylidium debile (150,105 bp) and Stylidium petiolare (150,998 bp). Both plastomes had the typical quadripartite structure, with large single-copy (LSC) and small single-copy (SSC) regions separated by two inverted repeat (IR) regions. Both plastomes have lost the rps19 and ycf15 CDS genes, and had infA-like, rps22-like, and rps7-like pseudogenes. Moreover, IR regions were expanded by having trnHGUG tRNA and the rps22-like pseudogene. Plastome phylogenomic analyses showed that the two Stylidium species formed a monophyletic clade (BS = 100), sister to the Argophyllaceae (BS = 86/83). Sequence differences between the two Stylidium plastomes were 5011 sites, including 2166 variable sites and 2845 indels, with the petA-psbJ spacer the most variable region, followed by the trnKUUU-matK intron and trnGUUG-rps16 spacer.
Stylidium (Stylidiaceae) are herbaceous perennials with the vernacular name of trigger-plants because of their pollination mechanism (Carolin Citation2007). There are around 220 species, mostly endemic to Australia, but with a few species in Southeast Asia and Sri Lanka, and also in New Zealand and southern South America (Carolin Citation2007). Some species are considered to be protocarnivorous (or subcarnivorous) because the glandular hairs trap insects (Darnowski et al. Citation2006). To date, phylogenetic relationships among the Stylidiaceae and within Stylidium are not well resolved (Laurent et al. Citation1998; Wagstaff and Juliet Citation2002), and only a few DNA regions from several species of Stylidium are deposited in GenBank, with no chloroplast genome sequences (accessed on 9 May 2021). In this study, we therefore sequenced and analyzed the complete chloroplast genome (or plastome) sequences of two Australian species, Stylidium debile F. Muell. 1859 and Stylidium petiolare Sond. 1845, which can be used to develop highly variable and informative plastid DNA regions for phylogenetic and population genetic studies of Stylidiaceae and Stylidium.
Genomic DNA of Stylidium debile and S. petiolare was extracted by a modified CTAB method (Doyle and Doyle Citation1987) from the silica-gel dried leaves, which were collected from cultivated living plants in the greenhouse of the Chinese Carnivorous Plants Garden (Chongfu Town, Tongxiang, Zhenjiang, China; 30°30′55.5″N, 120°24′46.5′E). The genomic DNA samples were then purified and fragmented into the size of 350 bp for preparing the sequencing library. The library preparation followed Zeng et al. (Citation2018). Around 2.0 GB clean data for each species were generated by Illumina Hi-Seq 2500 System via the 150 bp pair-end reads approach. Complete plastomes of these two species were de novo assembled by the GetOrganelle toolkit (Jin et al. Citation2020). The two plastome sequences were initially annotated by the GeSeq online tool (Tillich et al. Citation2017), then the start and stop codons were manually checked and adjusted by Geneious (Kearse et al. Citation2012). The voucher specimens (Stylidium debile: Liu G.-M. QT05; Stylidium petiolare: Liu G.-M. QT22) and DNA (Stylidium debile: Yu W.-B. P476; Stylidium petiolare: Yu W.-B. P475) are preserved in the herbarium of Xishuangbanna Tropical Botanical Garden, Chinese Academy of Sciences (HITBC, http://hitbc.xtbg.ac.cn/, Jian-Wu Li, [email protected]) and in the Germplasm Bank of Wild Species, Kunming Institute of Botany, Chinese Academy of Sciences (http://www.genobank.org/, Jun-Bo Yang, [email protected]), respectively.
The two plastome sequences had a typical quadripartite structure, with two inverted repeat (IR) regions separating large single-copy (LSC) and small single-copy (SSC) regions. The plastome sizes of Stylidium debile (accession number: MZ239203) and Stylidium petiolare (accession number: MZ239204) were 150,105 bp (GC content: 37.9%; LSC: 81,752 bp; SSC: 18,341 bp; IR: 25,006 bp) and 150,998 bp (GC content: 30.8%; LSC: 81,869 bp; SSC: 18,285 bp; IR: 25,422 bp), respectively. Both plastomes contained 131 genes in total, including 80 protein coding (CDS) genes, 38 tRNAs, eight rRNAs, and four pseudogenes (infA-like and rps22-like in LSC, and rps7-like in IR). The IR region had 19 genes, including six CDS genes, eight tRNAs, and four rRNAs, rps7-like pseudogene, and partial rps22-like pseudogene. Compared with the plastome of Amborella trichopoda Baill. (AJ506156), these two Stylidium plastomes had lost rps19 and ycf15 CDS genes, and their IR regions were expanded by having trnHGUG tRNA and an rps22-like pseudogene.
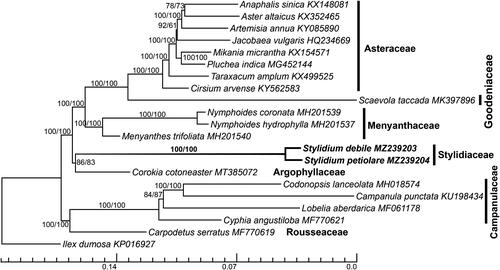
Twenty realigned plastomes of Asterales and one of Ilex dumosa Reissek (Aquifoliales, KP016927) as an outgroup with one IR region were aligned using MAFFT (Katoh and Standley Citation2013). Gaps in the initial matrix were removed by trimAl (Capella-Gutiérrez et al. Citation2009) using a command ‘-gt 0.9 -cons 60’. To infer the phylogeny of the Asterales, we performed maximum-likelihood analyses using RAxML (Stamatakis et al. Citation2008) by the GTR + GAMMA + I model with 1000 bootstraps for calculating support values for nodes/branches. The initial and trimmed matrixes were analyzed separately. Plastome phylogenies recovered the expected familial relationships among the Asterales (Stevens Citation2001; Li et al. Citation2019), with the exception that Argophyllaceae was sister to Stylidiaceae with moderate support values by initial (BS = 86) and trimmed datasets (BS = 83, see ). Moreover, Stylidium debile and Stylidium petiolare formed a monophyletic clade (BS = 100). There were 5011 sites with sequence differences, 2166 variable sites, and 2845 indels, between Stylidium debile and Stylidium petiolare. The petA-psbJ spacer was the most variable region, followed by the trnKUUU-matK intron (pi = 0.040–0.070) and the trnGUUG-rps16 spacer (pi = 0.037–0.070). This study provides important plastomic information and identifies hypervariable regions that can be used in future studies on the phylogeny and evolution of the genus Stylidium.
Figure 1. Plastome phylogenomics of Asterales inferred using the maximum-likelihood (ML) method. Stylidium debile and Stylidium petiolare are highlighted in bold. ML bootstrap values of nodes inferred using initial and trimmed matrixes, respectively, are presented next to the nodes. The bottom scale bar represents the number of substitutions per site.
Acknowledgements
We are grateful for the physical support from the HPC Platform of the Public Technology Service Center, Xishuangbanna Tropical Botanical Garden, Chinese Academy of Sciences, and The Molecular Biology Center, Germplasm Bank of Wild Species, Kunming Institute of Botany, Chinese Academy of Sciences.
Disclosure statement
The authors report no conflict of interest.
Data availability statement
Two complete plastome sequences were deposited in GenBank with accession numbers MZ239203 and MZ239204, and are also available at Figshare (doi:10.6084/m9.figshare.14649921). The associated BioProject, SRA, and Bio-Sample numbers are PRJNA739677, SRR14871484–SRR14871485, and SAMN19789662–SAMN19789663, respectively. The data were collected without violation of the protection of human subjects, or other valid ethical, privacy, or security concerns.
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article.
Additional information
Funding
References
- Capella-Gutiérrez S, Silla-Martínez JM, Gabaldón T. 2009. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics. 25(15):1972–1973.
- Carolin RC. 2007. Stylidiaceae. In: Kadereit JW, Jeffrey C, editors. Flowering plants. Eudicots Asterales. Berlin: Springer; p. 614–619.
- Darnowski DW, Carroll DM, Płachno B, Kabanoff E, Cinnamon E. 2006. Evidence of protocarnivory in triggerplants (Stylidium spp.; Stylidiaceae). Plant Biol. 8(6):805–812.
- Doyle JJ, Doyle JL. 1987. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochemistry. 19:11–15.
- Jin J-J, Yu W-B, Yang J-B, Song Y, dePamphilis CW, Yi T-S, Li D-Z. 2020. GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 21(1):241.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30(4):772–780.
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, et al. 2012. Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 28(12):1647–1649.
- Laurent N, Bremer B, Bremer K. 1998. Phylogeny and generic interrelationships of the Stylidiaceae (Asterales), with a possible extreme case of floral paedomorphosis. Syst Bot. 23(3):289–304.
- Li HT, Yi TS, Gao LM, Ma PF, Zhang T, Yang JB, Gitzendanner MA, Fritsch PW, Cai J, Luo Y, et al. 2019. Origin of angiosperms and the puzzle of the Jurassic gap. Nat Plants. 5(5):461–470.
- Stevens PF. 2001. Angiosperm phylogeny website. Version 12, July 2012 [and more or less continuously updated since]. http://www.mobot.org/MOBOT/Research/APweb/welcome.html.
- Stamatakis A, Hoover P, Rougemont J. 2008. A rapid bootstrap algorithm for the RAxML web servers. Syst Biol. 57(5):758–771.
- Tillich M, Lehwark P, Pellizzer T, Ulbricht-Jones ES, Fischer A, Bock R, Greiner S. 2017. GeSeq- versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 45(W1):W6–W11.
- Wagstaff SJ, Juliet W. 2002. Patterns of diversification in New Zealand Stylidiaceae. Am J Bot. 89(5):865–874.
- Zeng C-X, Hollingsworth PM, Yang J, He Z-S, Zhang Z-R, Li D-Z, Yang J-B. 2018. Genome skimming herbarium specimens for DNA barcoding and phylogenomics. Plant Methods. 14:43.