
Abstract
Primula stenocalyx Maxim. is a perennial herb with purple umbel flowers. This alpine plant can survive at altitudes of 2700–4300 m. To explore the chloroplast genome, total DNA was extracted from a sample and sequenced, the reads of the chloroplast genome were assembled and annotated. The chloroplast genome of this plant has a circular form with a length of 153,678 bp comprising a large single-copy region (LSC, 84,236 bp), a small single-copy region (SSC, 17,746 bp), and two inverted repeats (IRs, 25,848 bp). The genome has a GC content of 37%. A total of 133 genes were predicted to contain 88 encoded proteins, 8 rRNAs and 37 tRNAs. The evolutionary history indicates that P. stenocalyx was grouped within Primula and formed a clade with Primula knuthiana with a 100% bootstrap support value. The complete cp genome of P. stenocalyx can serve as a reference for future studies on molecular biology, evolution, population genetics, taxonomy and resource protection.
Primula stenocalyx Maxim., belonging to Primulaceae, is a perennial herb with rosette leaves, typically indistinct petioles, and little purple umbel flowers (Hu and Kelso Citation1996). This plant is always found on south-facing grassy slopes, ditches, and peat and is widely distributed in Gansu, Sichuan Province and the Qinghai-Tibetan Plateau in China. The plant can survive at altitudes of 2700–4300 m (Chen and Hu Citation1990). Elumeeva has reported the leaf functional traits of this plant (Elumeeva et al. Citation2015). Here, we will report the complete chloroplast (cp) genome and analyze its phylogenetic relationship with other related species.
Several samples were collected from the Qilian Mountains (36°52’46"N, 102°17’15"E, Huzhu County) in Qinghai Province, and a specimen was deposited at the College of Ecological Environment and Resources, Qinghai Nationalities University (https://shxy.qhmu.edu.cn/, Junqiao Li, email: [email protected]) under voucher number HCEERQNU-20200516001. Total DNA was extracted from the fresh leaves of a sample with a Rapid Plant Genomic DNA Isolation Kit (Sangon Biotech (Shanghai) Co., Ltd.). Paired-end libraries with an average length of 500 bp were constructed and sequenced on the Illumina HiSeq4000 platform (Sangon Biotech (Shanghai) Co., Ltd.). The complete cp genome was assembled via NOVOPlasty 3.7.2 (Dierckxsens et al. Citation2017) with Primula knuthiana (GenBank accession no. NC_039350.1) as the reference genome and annotated via PGA (Qu et al. Citation2019).
The complete cp genome of P. stenocalyx (GenBank accession no. MW678839.1) has a typical quadripartite form with a length of 153,678 bp and is composed of a large single-copy region (LSC, 84,236 bp), a small single-copy region (SSC, 17,746 bp), and two inverted repeats (IR, 25,848 bp). The genome has a GC content of 37%. A total of 133 genes were predicted for this cp genome, comprising 88 encoded proteins, 8 rRNAs and 37 tRNAs.
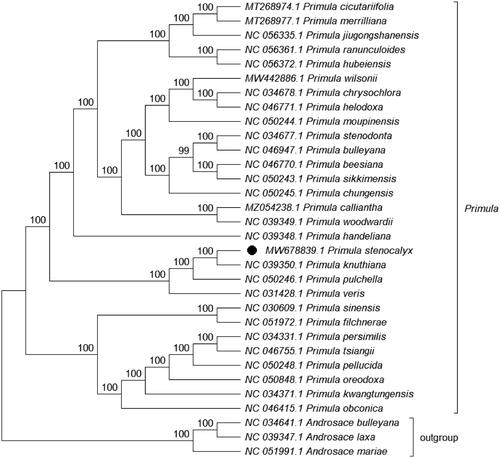
Phylogenetic analysis was performed on the complete cp genomes of P. stenocalyx and 28 other related species in Primula, with three species in Androsace as the outgroup. The genome-wide alignment was constructed by HomBlocks (Bi et al. Citation2018), the evolutionary history was inferred using the maximum likelihood (ML) method by IQ-TREE 1.6.12 with the TVM + F+R3 model (Nguyen et al. Citation2015; Kalyaanamoorthy et al. Citation2017), bootstrap (BS) values were calculated by UFBoot2 from 1000 replicates (Hoang et al. Citation2018), and the final output file was edited in MEGA X (Kumar et al. Citation2018). As expected, P. stenocalyx was grouped within Primula and formed a clade with Primula knuthiana with a 100% BS support value (). The complete cp genome of P. stenocalyx can serve as a reference for future studies on molecular biology, evolution, population genetics, taxonomy and resource protection.
Figure 1. ML phylogenetic tree based on 32 species chloroplast genomes was constructed using IQ-TREE 1.6.12. Numbers on each node are bootstrap support values from 1000 replicates.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The genome sequence data that support the findings of this study are openly available in GenBank of NCBI at (https://www.ncbi.nlm.nih.gov/nuccore/MW678839) under the accession no. MW678839.
The associated BioProject, SRA, and Bio-Sample numbers are PRJNA 725315, SRR 14361579, and SAMN 18875961, respectively.
Additional information
Funding
References
- Bi GQ, Mao YX, Xing QK, Cao M. 2018. HomBlocks: a multiple-alignment construction pipeline for organelle phylogenomics based on locally collinear block searching. Genomics. 110(1):18–22.
- Chen FH, Hu CM. 1990. Flora reipublicae popularis sinicae. Vol. 59. Beijing: Science Press; p. 193.
- Dierckxsens N, Mardulyn P, Smits G. 2017. NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 45(4):e18.doi:https://doi.org/10.1093/nar/gkw955. PMC: 28204566
- Elumeeva TG, Onipchenko VG, Yan W. 2015. Leaf functional traits of plants of alpine pastures at the Eastern Qinghai-Tibetan plateau. Moscow Univ Biolsci Bull. 70(1):46–52.
- Hoang DT, Chernomor O, von Haeseler A, Minh BQ, Vinh LS. 2018. UFBoot2: improving the ultrafast bootstrap approximation. Mol Biol E. 35(2):518–522.
- Hu CM, Kelso S. 1996. Flora of China. Vol. 15. Beijing: Science Press; p. 164.
- Kalyaanamoorthy S, Minh BQ, Wong TKF,von Haeseler A, Jermiin LS. 2017. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods. 14(6):587–589.
- Kumar S, Stecher G, Li M, Knyaz C, Tamura K. 2018. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 35(6):1547–1549.
- Nguyen LT, Schmidt HA, von Haeseler A, Minh BQ. 2015. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 32(1):268–274.
- Qu XJ, Moore MJ, Li DZ, Yi TS. 2019. PGA: a software package for rapid, accurate, and flexible batch annotation of plastomes. Plant Methods. 15:50.