
ABSTACT
Caroxylon passerinum is an important constructive species, which is widely distributed in both desert and desert steppe in north-western China. C. passerinum is one of hosts of holoparasitic Cistanche species. In this study, we report the complete chloroplast genome sequence of C. passerinum, which is 150,925 bp in length and comprises a large single-copy region (83,057 bp), a small single-copy region (18,180 bp), and a pair of inverted repeats (24,844 bp). It encodes 132 unique genes, including 89 protein-coding genes (PCGs), 35 tRNAs, and eight rRNAs. The overall GC content of this chloroplast genome is 36.8%. Maximum likelihood (ML) phylogenetic tree strongly supports that C. passerinum is closely related to the hosts of Cistanche deserticola, Haloxylon persicum and Haloxylon ammodendron.
Caroxylon passerinum (Chenopodiaceae) is an annual leaf-succulent and fruiting subshrub with extreme xerophyte and resistance to wind and cold (Akhani et al. Citation2007). Its populations are fragmentedly distributed in Inner Mongolia, Ningxia, Gansu and Xinjiang, as well as in southern Mongolia and Central Asia at elevations ranging from 1000 to 3000 m (Gao et al. Citation2009). As one of major constructive species, it is widely distributed in both desert and desert steppe in north-western China, with key ecological functions on sand fixation and preventing desertification (Reed Citation2003). C. passerinum and local medicine plants, the species of Cistanche such as Cistanche salsa, form a parasitism system (Qin and Liu Citation2010), which is a good system to study horizontal gene transfer (HGT) (Liu et al. Citation2020). In order to better understand its genomic structure and organization of C. passerimum, and its phylogenetic position in Chenopodiaceae, we sequenced the complete chloroplast genome of C. passerinum and compared with those of its relatives. The complete chloroplast genome of C. passerinum is the first report for any member of the genus Caroxylon.
We collected samples of C. passerinum in Zhongwei, Ningxia, China. The voucher specimens were deposited in the herbarium of Fudan University (FUS). Total genomic DNA was extracted from silica-gel dried leaves. Genomic DNA was sequenced using the Illumina Hiseq 2500 (Illumina, San Diego, CA, USA), with 150 bp paired-end (PE) sequencing. The chloroplast genome of Suaeda glauca (GenBank: MK867773) (Jian et al. Citation2019) served as our reference genome. Chloroplast assembling was carried out according to the previous method (Xu et al. Citation2020). The complete chloroplast genome was assembled by SOAPdenovo2 v2.04 (Luo et al. Citation2012) and Bowtie2 v2.3.4.1 (Langmead and Salzberg Citation2012). GapCloser v2.04 (Luo et al. Citation2012) was used to fill gaps among contigs. This draft genome was annotated using DOGMA (Wyman et al. Citation2004) and GeSeq (Tillich et al., Citation2017). To determine the phylogenetic placement of C. passerinum, a maximum-likelihood (ML) tree was reconstructed using RAxML v8.2.10 (Stamatakis Citation2014). tRNA genes were predicted using tRNAscan-SE v1.3.1 (Lowe and Chan Citation2016).
The chloroplast genome of C. passerinum is 150,925 bp in length (GenBank accession number: MW192441). It comprises a large single-copy region (LSC: 83,057 bp), a small single-copy region (SSC: 18,180 bp) and a pair of inverted repeats (IR: 24,844 bp). The overall GC content genome is 36.8%. A total of 132 unique genes were annotated, including 89 protein-coding genes, 35 tRNAs, and eight rRNAs.
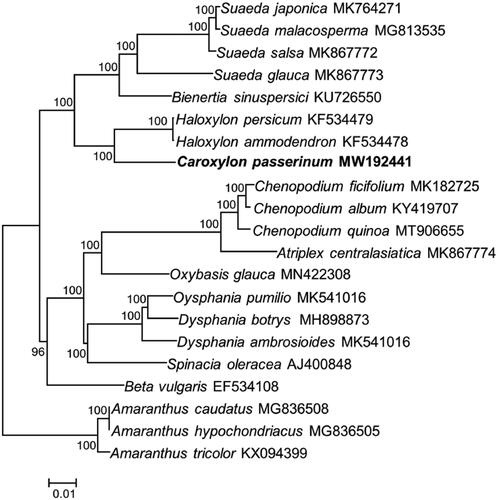
To validate the phylogenetic position of C. passerinum, 18 species from Chenopodiaceae and three species are from Amaranthaceae as complex outgroup were used to construct a ML phylogenetic tree. The sequences were aligned using MAFFT v7.309 (Katoh and Standley Citation2013) and the tree was analyzed with RAxML v8.2.11 (Stamatakis Citation2014). Phylogenetic analysis showed that C. passerinum is sister to members of Haloxylon. Besides, our results comfirmed that Caroxylon and Haloxylon had a closer relationship with Sueada and Bienertia than with Chenopodium ().
Figure 1. Molecular phylogeny of Chenopodiaceae using chloroplast genomes of 21 species including three species from Amaranthaceae as complex outgroup. Bootstrap values are based on 1000 replicates. The numbers on branches are bootstrap support values.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The data that support the findings of this study are openly available in NCBI GenBank at https://www.ncbi.nlm.nih.gov under the accession no. MW192441. The associated BioProject, SRA, and Bio-Sample numbers of Illumina raw sequencing data of Caroxylon passerinum are PRJNA673337, SRS7622123, and SAMN16604536, respectively.
Additional information
Funding
References
- Akhani H, Edwards G, Roalson EH. 2007. Diversification of the Old World Salsoleae s. l. (Chenopodiaceae): Molecular phylogenetic analysis of nuclear and chloroplast data sets and a revised classification. Int J Plant Sci. 168(6):931–956.
- Gao T, Gao H, Zhang Y, Xu Y, Li Z. 2009. Genetic diversity of Salsola passerina populations in northwestern China based on inter-simple sequence repeat (ISSR). J Lanzhou Univ Nat Sci. 45(2):66–74.
- Jian X, Liu L, Zhang L, Zhang X, Fan S. 2019. The complete chloroplast genome of an annual halophyte herb, Suaeda glauca (Amaranthaceae). Mitochondrial DNA B Resour. 4(2):2780–2781.
- Katoh K, Standley DM. 2013. MAFFT Multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30(4):772–780.
- Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods. 9(4):357–359.
- Liu X, Fu W, Tang Y, Zhang W, Song Z, Li L, Yang J, Ma H, Yang J, Zhou C, et al. 2020. Diverse trajectories of plastome degradation in holoparasitic Cistanche and genomic location of the lost plastid genes. J Exp Bot. 71(3):877–892.
- Lowe T, Chan P. 2016. tRNAscan-SE On-line: integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 44(W1):W54–W57.
- Luo R, Liu B, Xie Y, Li Z, Huang W, Yuan J, He G, Chen Y, Pan Q, Liu Y, et al. 2012. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience. 1(1):18.
- Qin H, Liu B. 2010. China checklist of higher plants. In the Biodiversity Committee of Chinese Academy of Sciences (ed.) Catalogue of Life China: 2010 Annual Checklist China. CD-ROM; Species 2000 China Node, Beijing, China.
- Reed D. 2003. Correlation between fitness and genetic diversity. Conserv Biol. 17(1):230–237.
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30(9):1312–1313.
- Tillich M, Lehwark P, Pellizzer T, Ulbricht-Jones ES, Fischer A, Bock R, Greiner S. 2017. GeSeq - versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 45(W1):W6–W11.
- Wyman SK, Jansen RK, Boore JL. 2004. Automatic annotation of organellar genomes with DOGMA. Bioinformatics. 20(17):3252–3255.
- Xu H, Fu W, Xie W, Wang Y, Zhang Y. 2020. The complete chloroplast genomes of two species of Zygophyllum (Zygophyllaceae). Mitochondrial DNA B Resour. 5(3):3476–3495.