
Abstract
Anopheles anthropophagus (Xu and Feng Citation1975) is the major vector of malaria in Eastern and Southern China. The species An. anthropophagus is considered a synonym of An. lesteri (Baisas & Hu, 1936), although they differ in several key biological characteristics. Here, we report the complete mitochondrial genome of An. anthropophagus for the first time. The mitogenome of An. anthropophagus is a typical circular, double-stranded molecule with a total length of 15,413 base pairs, and contains 13 protein-coding genes, 22 transfer RNA genes, two ribosomal RNA genes, and an AT-rich control region. A phylogenetic analysis of the complete mitogenomes of 16 species of Anopheles (Culicidae) revealed that An. anthropophagus is closely related to An. sinensis (Wiedemann 1828), in the family Culicidae. The An. anthropophagus mitogenome provides new data for further taxonomic and phylogenetic studies of the genus Anopheles.
Anopheles anthropophagus belongs to the Anopheles hyrcanus group (Xu and Feng Citation1975), which is widely distributed in Oriental and Palearctic regions. The species in the hyrcanus group are difficult to distinguish morphologically, especially An. lesteri and An. sinensis (Gao et al. Citation2004). The species An. anthropophagus is considered a synonym of An. Lesteri (Wilkerson et al. Citation2003; Rueda et al. Citation2005; Qu Citation2008), and has been identified as the main vector of malaria and Malayan filariasis in China (Zhu et al. Citation2013). After decades of effort to control malaria, especially vigorous vector control measures, the population of An. anthropophagus has been drastically reduced and has even disappeared in some areas, including provinces in Eastern and Southwestern China (Xu et al. Citation2001; Pan et al. Citation2012). On the contrary, the population of An. sinensis, which is sympatric with An. anthropophagus, has not declined to the same extent. Analysis of the mitochondrial genome of An. anthropophagus could provide insights that extend our understanding of the genetic characteristics and structure of the genus Anopheles (Cameron Citation2014).
In this study, we determined the complete mitochondrial genome of An. anthropophagus (Genbank: MW279150) for the first time and analyzed it phylogenetically. The specimens of An. anthropophagus was originally collected in the north of Jiangsu Province, reared in the laboratory of Jiangsu Institute of Parasitic Diseases, Wuxi, Jiangsu, China (31°56′N,120°23′E), and were deposited at Anhui Provincial Key Laboratory of Infection and Immunology, Bengbu Medical College, Bengbu, Anhui, China (voucher Mos-2020-An-001, Baiqing Li, [email protected]). DNA was extracted from the mosquito tissue with the AxyPrep Plasmid Miniprep Kit (Axygen, China). To amplify the entire mitochondrial genome of An. anthropophagus, 11 pairs of primers (Supplementary Table S1) were designed based on the known mitogenomes of mosquito species. The PCR products were sequenced using the Sanger sequencing method (Sangon Biotech, China). The raw nucleotide sequences were trimmed to remove low-quality ends and assembled with DNASTAR V11.0 Lasergene (DNA Star). MITOS (http://mitos2.bioinf.uni-leipzig.de) (Bernt et al. Citation2013) was used to annotate the mitogenome.
The mitochondrial genome of An. anthropophagus is a typical circular, double-stranded molecule with a total length of 15,413 base pairs, and contains 13 protein-coding genes, 22 transfer RNA genes, two ribosomal RNA genes, and a control region. The nucleotide composition of the mitochondrial genome is 40.21% A, 38.03% T, 9.20% G, and 12.56% C. The A + T base composition (78.24%) of the complete mitogenome is higher than the G + C base composition (21.76%), and thus, the nucleotide composition is highly A + T biased. The locations and order of all genes are similar to those in other Anopheles species (Chen et al. Citation2017). The start codon ATG is used by six protein-coding genes (COII, ATP6, COIII, ND4, ND4L, and CYTB), ATT by four protein-coding genes (ND2, ATP8, ND6, and ND1), TCG by COI, ATA by ND3, and GTG by ND5. All 13 protein-coding genes use either TAA (ND2, ATP8, ATP6, ND3, ND4L, ND5, ND6, Cyt B, ND1) or T (CO1, CO2, CO3, ND4) as the termination codon. The 22 tRNAs range in length from 62 to 72 bp with a typical cloverleaf structure. The mitogenome of An. anthropophagus contains two rRNA genes, 16S rRNA and 12S rRNA, they are all located in the N chain. The 16S rRNA is 1,335 in length and is located between tRNALeu and tRNAVal. The 12S rRNA is 794 bp in length and is located between tRNAVal and the control region. The length of the control region is 566 bp with high A + T content (94.17%), and is located between 12S rRNA and tRNAIle. 9 intergenic spacer regions are ranging in length from 1 to 20 bp, and the largest interval appears between tRNASer and ND1. Thirteen intergenic overlapping regions are between 1 and 7 bp in size (Supplementary Table S2).
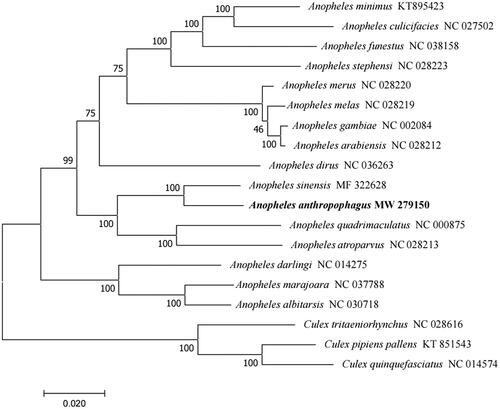
The phylogenetic analysis of the complete mitochondrial genomes of Anopheles species was performed with the genus Culex as the outgroup. A phylogenetic tree was constructed using the maximum likelihood (ML) method in MEGA 7.0 (Sudhir et al. Citation2016) under the Tamura-Nei model, with 1000 bootstrap replicates. The results showed that An. anthropophagus is closely related to An. sinensis (), which are sympatric species and have both been confirmed as the major vectors of Plasmodium vivax in China. In conclusion, the complete mitochondrial genome of An. anthropophagus reported in this study provides useful information for further taxonomic, phylogenetic, and evolutionary analyses of the genus Anopheles.
Supplemental Material
Download MS Word (21.1 KB)Acknowledgments
The authors thank Dr. Qi Gao and Dr. Jun Cao of Jiangsu Institute of Parasitic Diseases for helpful discussion.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The genome sequence data that support the findings of this study are openly available in GenBank of NCBI at (https://www.ncbi.nlm.nih.gov/)under the accession no. MW279150.
Figure 1. Phylogenetic tree of 16 Anopheles species based on their mitochondrial genomes. Numbers near the nodes represent bootstrap values. The GenBank accession number is listed next to each species within the tree. Culex was used as the outgroup. The branch length scale bar indicates relative differences (0.020 = 2.0% nucleotide difference).
Additional information
Funding
References
- Bernt M, Donath A, Juhling F, Externbrink F, Florentz C, Fritzsch G, Putz J, Middendorf M, Stadler PF. 2013. MITOS: improved de novo metazoan mitochondrial genome annotation. Mol Phylogenet Evol. 69(2):313–319.
- Cameron SL. 2014. Insect mitochondrial genomics: implications for evolution and phylogeny. Annu Rev Entomol. 59:95–117.
- Chen K, Wang Y, Li X-Y, Peng H, Ma Y-J. 2017. Sequencing and analysis of the complete mitochondrial genome in Anopheles sinensis (Diptera: Culicidae). Infect Dis Poverty. 6(1):149 doi:https://doi.org/10.1186/s40249-017-0362-7. PMC: 28969698
- Gao Q, Beebe NW, Cooper RD. 2004. Molecular Identification of the malaria vectors Anopheles anthropophagus and Anopheles sinensis (Diptera: Culicidae) in central China using polymerase chain reaction and appraisal of their position within the Hyrcanus group. J Med Entomol. 41(1):5–11.
- Pan JY, Zheng X, Qian HL, Liu CF, Guo CK, Jiang MG, Chen HL, Gu ZC. 2012. Impact of pesticide use on the density of Anopheles anthropophagus and malaria incidence. Zhongguo Ji Sheng Chong Xue Yu Ji Sheng Chong Bing Za Zhi. 30(3):218–223. in Chinese)
- Qu FY. 2008. Historical review on the classification and rectification of Anopheles anthropophagus to An. lesteri in China. Zhongguo Ji Sheng Chong Xue Yu Ji Sheng Chong Bing Za Zhi. 26(3):234–235.
- Rueda LM, Wilkerson RC, Li C. 2005. Anopheles (Anopheles) lesteri Biases and Hu (Diptera: Culicidae): neotype designation and description. Proc Entomol Soc Wash. 107(3):604–622.
- Sudhir K, Glen S, Koichiro T. 2016. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol E. 33(7):1870–1874.
- Wilkerson RC, Li C, Rueda LM, Kim HC, Klein TA, Song GH, Strickman D. 2003. Molecular confirmation of Anopheles (Anopheles) lesteri from the Republic of South Korea and its genetic identity with An. (Ano.) anthropophagus from China (Diptera: Culicidae). Zootaxa. 378(1):1–14.
- Xu JJ, Feng LZ. 1975. Studies on the Anopheles Hyrcanus Group of mosquitoes in China. Acta Entomologica Sinica. 18(1):77–98.
- Xu HB, Xu LS, Li LS. 2001. The effect of the surviving Anopheles anthropophagus eradicated in Fujian province. Chinese J Vector Biol Control. 12(6):421–423.
- Zhu G, Xia H, Zhou H, Li J, Lu F, Liu Y, Cao J, Gao Q, Sattabongkot J. 2013. Susceptibility of Anopheles sinensis to Plasmodium vivax in malarial outbreak areas of central China. Parasit Vectors. 06(1):176.